
Necrostatin-1对TLR4/NF-κB途径的调控在大鼠脑缺血再灌注后小胶质细胞活化中的意义
2019-01-11 07:56刘芷含张瑞成周延龙耿德勤程言博
中风与神经疾病杂志 2018年12期
杨 龙, 刘芷含, 张瑞成, 冯 宇, 周延龙, 贾 潇, 耿德勤, 程言博
小胶质细胞(MG)的活化是中枢神经系统触发炎症反应的主要因素[1]。脑缺血以后,小胶质细胞被活化,成为一把“双刃剑”[2],起损伤和修复双重效应,因此得到广泛的关注。程序性坏死(Necroptosis)是一种caspases非依赖的细胞死亡方式,能被受体作用蛋白-1(RIP1)的别构抑制剂Nec-1所抑制[3],Nec-1参与活化的小胶质细胞的调控,能通过影响炎症信号途径而发挥抗炎作用[4,5]。我们前期研究发现,Nec-1能使大鼠脑缺血再灌注时,炎症因子如白细胞介素-1β(IL-1β)和肿瘤坏死因子-α(TNF-α)产生明显减少[6]。但Nec-1在减轻缺血再灌注损伤进程中炎症反应的具体信号通路及其调控机制尚不清楚。研究表明在MCAO模型中,TLR4、NF-κB和IL-6参与脑缺血再灌注损伤的过程[7,8]。Nec-1减轻脑缺血再灌注损伤后炎症反应和小胶质细胞活化,是否与TLR4/NF-κB途径以及IL-6有关,目前文献少有报道。因此,本研究拟通过线栓法制备大鼠MCAO模型,通过检测小胶质细胞标记物iba-1以及TLR4、NF-κB和IL-6蛋白表达情况,探讨Nec-1减轻大鼠脑缺血再灌注损伤后炎症反应和小胶质细胞活化的可能机制,为缺血性脑血管病的治疗提供新的靶点和思路。
1 材料和方法
1.1 材料
1.1.1 实验动物 成年雄性SD大鼠96只,清洁级,体重(250±10)g,由徐州医科大学实验动物中心提供,许可证号:苏SYXK2002-0038。
1.1.2 主要实验试剂及材料 MCAO栓线(北京沙东生物技术公司),Necrostatin-1(Selleckchem 公司),羊多克隆抗iba-1抗体、小鼠单克隆抗TLR4抗体(Abcam公司),兔多克隆抗IL-6抗体(药科美生物技术有限公司),兔多克隆抗NF-κB/p65抗体(CST公司)。
1.2 方法
1.2.1 取SD大鼠48只,体重(250±10)g,按随机数表法随机分为Sham组、MCAO组、DMSO组、Nec-1干预组,每组再分两个亚组,分别为再灌注6 h、24 h,每组6只。Nec-1组于缺血前30 min按640 nmol/kg侧脑室注射Nec-1溶液(溶于10%DMSO溶液),DMSO组注射等量DMSO溶液。线栓法制备MCAO模型,于缺血2 h再灌注6 h和24 h,免疫组化染色检测TLR4、NF-κB/p65和iba-1的免疫阳性细胞表达。
1.2.2 另取48只SD大鼠,随机分为Sham组、MCAO组、DMSO组、Nec-1干预组,每组再分两个亚组,每组6只,于缺血2 h、再灌注6 h和24 h,WB检测TLR4、NF-κB/p65、IL-6蛋白的定位和表达。
1.3 MCAO模型的建立 根据Longa线栓法制备MCAO模型[9]。将大鼠麻醉后固定,分离右侧颈动脉。结扎颈总动脉近心端,远心端用动脉夹阻断血流,两者之间剪一V形小口。将MCAO栓线沿V形口插人颈总动脉,去掉动脉夹,将栓线缓缓推入至右侧大脑中动脉起始处,进线深度约18~20 mm。结扎远心端固定栓线,常规消毒缝合切口,缺血2 h后拔除栓线行再灌注。
1.4 侧脑室给药参照文献方法[10]。
1.5 神经功能学评分及纳入排除标准 (1)根据Longa方法[9]对脑缺血后2 h的大鼠进行神经功能评分。自由活动,没有神经功能障碍者,0分;提起大鼠尾巴左前肢屈曲者,1分;行走时地上打转,静止时身体无偏向左侧者,2分;行走时地上打转,静止时身体也偏向左侧者,3分;不能行走,严重意识障碍者,4分。(2)纳入及排除标准:评分在1~3分的大鼠纳入研究。因麻醉原因、手术操作及蛛网膜下腔出血造成的大鼠死亡和评分低于1分的大鼠予以排除,并随机选取相同条件大鼠补充。

2 结 果
2.1 免疫组织化学染色 与Sham组相比,MCAO组梗死周围组织iba-1、TLR4、NF-κB/p65阳性细胞明显增多,差异有统计学意义(P<0.05);与 MCAO组相比,Nec-1组其阳性细胞明显减少,差异有统计学意义(P<0.05);DMSO组与MCAO组相比差异无统计学意义(P>0.05)(见图1、表1、表2)。
2.2 Western blot结果 通过分析灰度值,Sham组梗死周围组织有少量TLR4、IL-6表达,细胞核内有NF-κB/p65表达较低,MCAO组大鼠TLR4、IL-6和核内NF-κB/p65的表达显著增加;与MCAO组相比,Nec-1组TLR4、IL-6的表达量显著减少,核内NF-κB/p65的表达量显著减少,核转位减轻,差异有统计学意义(P<0.05);DMSO组与MCAO组相比差异无统计学意义(P>0.05)(见图2、图3)。
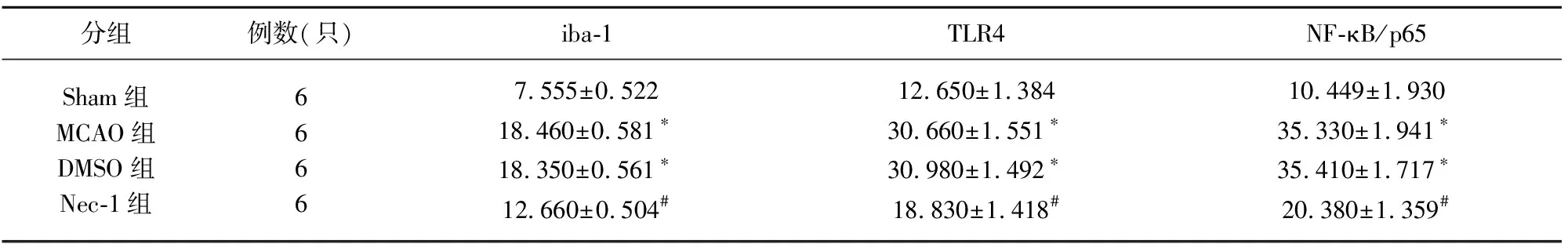
表1 I/R后6 h各组大鼠iba-1、TLR4、NF-κB/p65免疫阳性细胞表达值比较(×10-3/μm2)
与Sham组相比*P<0.05;与MCAO组相比#P<0.05

表2 I/R后24 h各组大鼠iba-1、TLR4、NF-κB/p65免疫阳性细胞表达值比较(×10-3/μm2)
与Sham组相比*P<0.05;与MCAO组相比#P<0.05
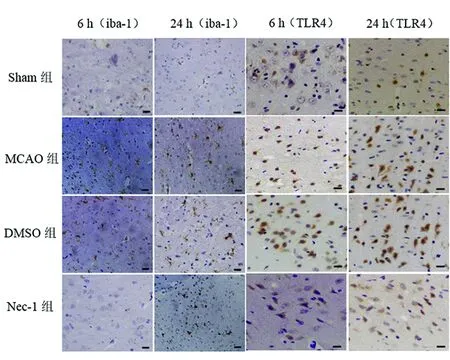
图1 I/R后 6 h和24 h梗死周围组织iba-1和TLR4免疫组化染色(Bar=20 μm,×400)
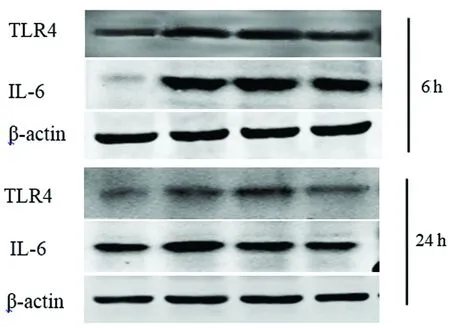
图2 I/R后 6 h和24 h梗死周围组织TLR4、IL-6蛋白表达结果
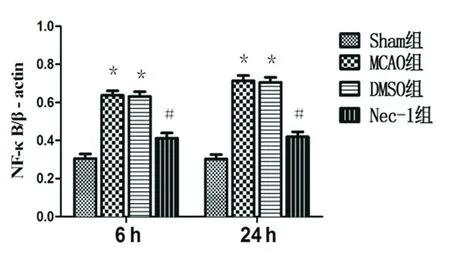
与Sham相比*P<0.05;与MCAO组相比#P<0.05
图3 I/R6 h和24 h梗死周围组织胞核内NF-κB/p65蛋白灰度值比较
3 讨 论
小胶质细胞的活化是中枢神经系统触发炎症反应的主要因素[1,4,5]。当脑缺血再灌注时,死亡的神经细胞会释放热休克蛋白-60、透明质酸、纤维蛋白原等内源性配体,位与小胶质细胞表面的TLR4则能结合这些配体,导致小胶质细胞活化[11],活化后会释放大量炎性介质,引起继发性神经元损伤,从而形成恶性循环造成级联放大效应,加重脑组织损伤。脑组织在发生可逆性缺血事件损伤后,又遭遇了不可逆的二次打击:小胶质细胞的过度活化和炎性因子的大量释放[12]。
正是由于缺血事件中的二次打击,寻找一种新的药物来抑制小胶质细胞的活化,减轻炎症反应显得尤为重要。抑制caspases活性可以通过靶向作用于小胶质细胞来起神经保护作用[13],而程序性坏死是caspases非依赖的细胞死亡方式。有证据表明,体内缺失受体相互作用蛋白3,则炎症反应消失,说明炎症反应可能源自程序性坏死[14],且前期研究也发现脑缺血再灌注后,给予Nec-1干预,炎性因子产生明显减少[4,6,15],对脑组织起保护作用,这也对干预程序性坏死途径抗炎治疗提供了可能的依据。
RIP1是程序性坏死的关键激酶,其激酶活性对细胞坏死和凋亡起关键作用[16];而Nec-1通过抑制RIP1激酶活性来保护神经组织,可能与减轻下游炎症反应有关[15],在脑I/R模型中,Nec-1是通过何种途径减轻炎症反应和小胶质细胞活化来保护神经组织,目前尚无确切研究结果。
有研究指出,RIP1是TLR3激活NF-κB信号通路中的基本介导物[17],但RIP1是否在TLR4激活NF-κB通路中起重要作用,目前文献鲜有报道。因此本研究,通过观察Nec-1对大鼠I/R后TLR4、NF-κB/p65、IL-6的表达的影响,探讨Nec-1在大鼠I/R后小胶质细胞活化中的意义,和可能的脑保护机制。
TLR4途径对小胶质细胞活化和中枢神经系统炎症反应调控居于核心地位,在脑内主要由小胶质细胞表达。脑缺血时,TLR4基因敲除的小鼠炎性因子产生明显减少,脑组织损伤减轻[7]。TLR4 激活后主要通过两条信号通路产生作用:髓样分化因子88(MyD88)依赖信号通路和非MyD88依赖信号通路。MyD88依赖信号通路主要作用于NF-κB,其活化后可以促使TNF-α、IL-6等炎性介质的分泌,引起免疫炎症反应[18]。问题是脑I/R时,活化的小胶质细胞调节TNF-α、IL-6等炎症因子的表达是否通过对TLR4、NF-κB的调控而实现的?
我们前期研究发现,脑缺血再灌注6 h后小胶质细胞开始出现活化,至24 h后活化达到高峰[19]。因此本研究选择再灌注6 h和24 h两个时间点。通过免疫组化和WB观察到假手术组大鼠TLR4和核内NF-κB表达极低,手术组表达明显增高,证明TLR4/NF-κB信号途径在大鼠缺血性脑损伤中起重要作用。TLR4升高的可能机制为:组织因缺血缺氧坏死产生的大量内源性配体如热休克蛋白60、透明质酸等,与TLR4结合,导致小胶质细胞活化,后者释放大量炎性介质,作用于临近的小胶质细胞,引起中枢神经炎症与胶质细胞激活的反馈环,从而放大神经毒性分子的产生引起继发性神经元损害;受损的神经元因为这个二次打击释放更多的内源性配体激活TLR4。Nec-1干预组较手术组表达降低,考虑可能机制有:Nec-1拯救死亡脑组织,使组织程序性坏死减少,小胶质细胞表面TLR4内源性配体减少;IL-6和TNF-α等促炎性因子表达减少,促炎性因子刺激新生小胶质细胞活化减少,使炎症反应减轻。
此外,本研究还观察到在缺血2 h,再灌注24 h,Nec-1预处理能明显改善神经功能缺损症状,TTC染色显示脑梗死体积减小,并使脑水肿程度明显减轻。因此我们可以推断,在大鼠I/R损伤模型中,Nec-1通过拯救缺血脑组织的程序性坏死,使小胶质细胞表面TLR4受体激活减少,至下游经典转录因子NF-κB核移位减少,从而减轻脑缺血再灌注损伤后的炎症反应,对脑组织起保护作用。活化的小胶质细胞通过necroptosis途径分泌TNF-α、IL-6等因子可能是通过对TLR4、NF-κB的调控而实现的。
综上所述,尽管目前已发现程序性坏死某些调控炎症的分子机制,但具体的信号机制尚不明确,TLR4信号通路介导的小胶质细胞活化和炎症因子的释放是其重要机制之一。动物在体模型中,TLR4、RIP1和NF-κB可能不是简单的上游与下游的关系,他们可能在促炎症因子IL-6等的参与下,形成复杂交互的网络关系,起到相互调节的作用,RIP1在炎症反应中起中枢调控作用[20]。对TLR4及其信号通路进行干预,可能会为人类治疗缺血性脑血管病后炎症反应提供新的思路。但是,本研究仅局限于动物在体模型中,另外没有观察对其他炎性因子和活性物质表达的影响,这些均需要进一步深入细致研究。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
天津医科大学学报(2021年3期)2021-07-21
昆明医科大学学报(2020年11期)2020-12-28
神经损伤与功能重建(2020年10期)2020-12-23
神经损伤与功能重建(2020年11期)2020-12-01
意林·少年版(2019年11期)2019-06-30
科学之谜(2019年3期)2019-03-28
飞碟探索(2015年11期)2015-09-10
