
别隐品碱衍生物的合成及其生物活性
2018-12-06 03:51李浩鹏史晨燕刘睿媛耿会玲
西北农林科技大学学报(自然科学版) 2018年11期
李浩鹏,史晨燕,刘睿媛,耿会玲
(西北农林科技大学 化学与药学院,陕西 杨凌 712100)
别隐品碱对肿瘤细胞和恶性癌变细胞具有细胞毒活性和抑制细胞增殖活性,但比抗肿瘤药物奥沙利铂(oxaliplatine)、长春花碱(vinblastine)和环磷酰胺(cyclophosphamide)的效果稍差[1]。Sener和Orhan[2]研究了20多种球果紫蓳属植物提取物(主要成分为原阿片碱和别隐品碱)对乙酰胆碱酯酶的抑制活性发现,其抗乙酰胆碱酯酶作用具有可逆性和竞争性,且与剂量有关。目前临床上使用的许多药物都是在天然产物结构基础上经过修饰改造获得的模仿和优化产物[3]。对1981-2006年上半年全球上市新药来源的统计结果显示,所有药物中小分子新化合物实体药物占82%,其中6%直接来源于天然产物,36%来源于全合成;其余58%介于两者之间,包括天然产物衍生物、类似物、模拟物等[4]。可见对新药研发而言,天然产物的结构修饰是极其重要的内容。
别隐品碱是普罗托品类生物碱中的一种,普罗托品类生物碱具有多种药理活性,目前对其作用机理的研究也越来越深入。胡海军等[5]研究了血根碱和白屈菜红碱杀螨活性的构效关系,确定了此类化合物的活性部位是C=N+离子型双键。通过对普罗托品类生物碱的羰基和N进行初步结构修饰及杀螨活性构效关系的研究,推测14-位的羰基不是活性基团,但将其转化为羟基后活性大大提高;N不是活性部位,将其甲基化或氮氧化后所得衍生物的杀兔痒螨活性均无明显变化[6-9]。为了更系统地研究别隐品碱类生物碱的杀螨活性构效关系,进一步寻找杀螨活性药物,本试验以别隐品碱为原药,对其苯环和十元氮杂环进行结构修饰与改造及杀螨活性测定,分析其杀螨活性的构效关系,以期为后续结构优化和药品研发提供依据。
1 材料与方法
1.1 材 料
1.1.1 供试病原菌 供试苹果腐烂病菌(Valsamali)、番茄早疫病菌(Alternariasolani)、小麦赤霉病菌(Fusariumgraminearum)、玉米弯孢病菌(Curvularialunata)、马铃薯干腐病菌(Fusariumsolani)、水稻稻瘟病菌(Pyriculariaoryza),由西北农林科技大学无公害农药研究中心提供。
兔痒螨采自患有螨病的兔子(2015年11月购自陕西省周至县哑柏镇),经西北农林科技大学宋晓平教授鉴定为痒螨科(Psoroptidae)兔痒螨(P.cuniculi)。
1.1.2 仪器与试剂 XT-4显微熔点仪(北京泰克仪器有限公司),Bruker AVANCE Ⅲ 400 MHz核磁共振仪(德国Bruker公司),Finnigan Trace 质谱仪(美国Themo Finnigan公司)。
原药别隐品碱为在博落回中分离所得;98.5%嘧菌酯(Azoxystrobin)原药(北京颖泰嘉和科技有限公司产品);柱层析硅胶,0.050~0.074 mm(青岛海洋化工厂产品);用于分离纯化的试剂为重蒸的工业试剂,甲醇、无水乙醇和二氯甲烷等试剂均为市售分析纯试剂,必要时处理后使用。
1.2 目标化合物的合成
以别隐品碱(A)为原药,参考文献报道方法合成12种目标化合物,合成路线如图1所示。
1.2.1 化合物A1 依次将185 mg别隐品碱(0.5 mmol)和10 mL 干燥氯仿加入50 mL圆底烧瓶中,再在剧烈搅拌下缓慢滴加0.5 mL草酰氯((COCl)2),回流2 h后产生黄色沉淀,TLC检测显示反应完全。将反应液冷却至室温,减压抽滤得黄色粗产物[10],用乙醚洗涤3次(每次3~5 mL),抽干,得淡黄色粉末即化合物A1(167 mg),产率86%。
1.2.2 化合物A2 依次将148 mg别隐品碱(0.4 mmol)和8 mL乙酸加入25 mL圆底烧瓶中,N2保护下加入4 mL新配制的溴素乙酸溶液(19.7 mg/mL,Br2-AcOH)。室温搅拌1 h,有黄色固体生成[11]。减压抽滤,乙酸洗涤2次(每次3~5 mL)。滤液置换N2后再加入2 mL溴素乙酸溶液,减压抽滤,合并黄色固体[12]。硅胶柱层析(V乙醚∶V乙醇=1∶1)分离纯化得白色粉末状化合物A2(48 mg),产率27%。
1.2.3 化合物A3 分别将185 mg别隐品碱(0.5 mmol)和20 mL乙酸加入100 mL圆底烧瓶中,室温搅拌下,缓慢滴加5 mL新配制的HNO3-AcOH(二者体积比3∶2)混合溶液。反应15 min后,溶液变成红棕色,TLC检测显示反应完全[13-14],停止反应。将反应液倒入50 mL冰水中,用饱和NaHCO3溶液调pH至中性。用二氯甲烷萃取(15 mL×2),合并有机相,用饱和食盐水洗3次,无水Na2SO4干燥,硅胶柱层析(V二氯甲烷∶V甲醇=10∶1)分离纯化得淡黄色结晶即化合物A3(80 mg),产率38%。

a.(COCl)2,CHCl3,reflux;b.Br2-AcOH,N2;c.HNO3-AcOH(3∶2),15 min;d.I2,AcONa,EtOH,reflux;e.m-CPBA,0 ℃;f.LiAlH4,THF;g.m-CPBA,0 ℃;h.CH3I,CHCl3;i.NaH(65%),CH3I,THF;j.NaH(65%),CH3I,THF,40 ℃;k.CH3COCl;l.NaH(65%),CH3CH2I,THF
1.2.4 化合物A4 分别将370 mg别隐品碱(1 mmol)和60 mL无水乙醇加入150 mL圆底烧瓶,60 ℃加热搅拌溶解后,滴加10 mL的I2溶液(0.5 mol/L,溶剂为乙醇)。回流反应1 h,向烧瓶中加入3 mL醋酸钠溶液(AcONa),继续反应2 h,TLC检测显示反应完全,停止反应[15]。蒸干溶剂,加入40 mL二氯甲烷溶解,再加入30 mL饱和NaHSO3水溶液,静置分液。水相用二氯甲烷萃取(15 mL×2),合并有机相,饱和NaHCO3水溶液洗1次,饱和食盐水洗3次,无水Na2SO4干燥,硅胶柱层析(V二氯甲烷∶V甲醇=5∶1)分离纯化得无色结晶即化合物A4(200 mg),产率52%。
1.2.5 化合物A5 在100 mL圆底烧瓶中分别加入185 mg(0.5 mmol)别隐品碱和20 mL干燥的氯仿,溶解完全后在冰浴中搅拌0.5 h,缓慢加入10 mL间氯过氧苯甲酸钠(m-CPBA,体积分数75%)的氯仿溶液,-18 ℃下搅拌反应25 h,TLC检测反应完全。边搅拌边向反应液中缓慢加入20 mL 体积分数5%的Na2CO3溶液,氯仿萃取(30 mL×3),合并有机相,无水Na2SO4干燥,硅胶柱层析(V二氯甲烷∶V甲醇=10∶1)分离纯化得白色粉末即化合物A5(169 mg),产率89%。
1.2.6 还原产物A6 将185 mg(0.5 mmol)别隐品碱加入250 mL圆底烧瓶中,加入约60 mL无水四氢呋喃(THF),加热(40 ℃)溶解完全,冷却至室温后,边搅拌边缓慢加入40 mL用无水THF溶解的2.5 mmol氢化铝锂(LiAlH4)。8 h后TLC检测反应完全[16-17]。边搅拌边向反应液中逐滴加入约2 mL甲醇至溶液不再产生气泡,再加入2 mL体积分数10%的NaOH溶液,溶液中出现白色絮状沉淀,过滤,将滤液减压蒸除溶剂,得粗产物。硅胶柱层析(V石油醚∶V乙酸乙酯=5∶1)分离纯化得到无色晶体即化合物A6(159 mg),产率86%。
1.2.7 化合物A7 将186 mg(0.5 mmol)化合物A6和20 mL干燥氯仿加入100 mL圆底烧瓶中,加热(40 ℃)溶解完全。冷却至室温,在冰浴中搅拌0.5 h后缓慢加入10 mL含间氯过氧苯甲酸钠(m-CPBA,体积分数75%)的氯仿溶液, -18 ℃下搅拌反应25 h后,TLC检测反应完全[18]。边搅拌边向反应液中缓慢加入20 mL体积分数5% 的Na2CO3溶液,氯仿萃取(30 mL×3),合并有机相,无水Na2SO4干燥,过滤,减压浓缩有机相得粗产物。硅胶柱层析(V氯仿∶V甲醇=10∶1)分离纯化得白色粉末即化合物A7(165 mg),产率80%。
1.2.8 化合物A8 依次将185 mg(0.5 mmol)化合物A6和40 mL干燥氯仿加入100 mL圆底烧瓶中,超声并加热溶解完全。冷却至室温后,加入2 mL CH3I,室温下搅拌,溶液中缓慢出现白色不溶物,每隔24 h抽滤不溶物同时补加1 mL CH3I,TLC检测反应完全[19-20]。减压抽滤,用无水乙醇冲洗(15 mL×2),烘干后得白色粉末即化合物A8(165 mg),产率80%。
1.2.9 化合物A9 向100 mL圆底烧瓶中加入37 mg(0.1 mmol)化合物A6和10 mL 干燥的THF,室温搅拌下,加入约5 mg体积分数65%的NaH,至不再产生气泡。5 min后向反应瓶中缓慢加入2 mL CH3I,常温搅拌反应20 h后TLC检测反应完全,用约3 mL甲醇淬灭反应。溶液加压浓缩至干后加入10 mL CH2Cl2溶解,再加入10 mL水,用氯仿萃取(10 mL×3),合并有机相,用适量无水Na2SO4干燥,硅胶柱层析(V氯仿∶V甲醇=10∶1)分离纯化得无色晶体即化合物A9(28 mg),产率78%。
1.2.10 化合物A10 依次将186 mg(0.5 mmol)化合物A6和15 mL干燥THF加入100 mL圆底烧瓶中,完全溶解后,搅拌下分批加入0.2 g体积分数65%的NaH。80 ℃加热回流1 h后,得白色悬浮溶液,降温至40 ℃。将2 mL干燥碘甲烷缓慢滴加入上述反应液中,8 h后TLC检测原料反应完全。加入10 mL甲醇淬灭反应,减压蒸干溶剂,加入约40 mL水和40 mL二氯甲烷搅拌溶解,静置分液。水相用二氯甲烷萃取(20 mL×2),合并有机相,饱和食盐水洗2次,无水Na2SO4干燥,硅胶柱层析(V二氯甲烷∶V甲醇=8∶1)分离纯化得白色粉末即化合物A10(140 mg),产率78%。
1.2.11 化合物A11 依次将100 mg(0.27 mmol)化合物A6和7 mL干燥CH2Cl2加入50 mL圆底烧瓶中,缓慢滴加4 mL乙酰氯(CH3COCl),室温反应16 h,TCL检测反应完全。加入10 mL H2O水解多余的乙酰氯,饱和NaHCO3溶液调节pH至中性,补加15 mL水和15 mL二氯甲烷,静置分液。水相用二氯甲烷萃取(15 mL×2),合并有机相,饱和食盐水洗2次,无水Na2SO4干燥,蒸干溶剂得粗品[21]。粗品用甲醇/乙醚(V甲醇∶V乙醚=1∶1)重结晶,抽滤,减压烘干,得白色固体即化合物A11(71 mg),产率75%。
1.2.12 化合物A12 依次将186 mg(0.5 mmol)化合物A6和15 mL干燥THF加入100 mL圆底烧瓶中,搅拌下分批加入0.2 g NaH(3 mmol),80 ℃ 加热回流1 h后,得白色悬浮溶液,冷却至室温。将1.5 mL干燥碘乙烷溶于5 mL干燥四氢呋喃中稀释后,分3次加入上述反应液中, 24 h后TLC检测原料反应完全[22-23]。加入10 mL甲醇淬灭反应,减压蒸干溶剂,加入约40 mL水和40 mL二氯甲烷搅拌溶解,静置分液。水相用二氯甲烷萃取(20 mL×2),合并有机相,饱和食盐水洗2次,无水Na2SO4干燥,硅胶柱层析(V石油醚∶V乙酸乙酯=5∶1)分离纯化得白色固体即化合物A12(130 mg),产率70%。
1.3 化合物结构表征
采用质谱(ESI-MS)和核磁共振谱(1H NMR、13C NMR)等分析手段,对化合物A1~A12的结构进行表征。
1.4 化合物A1~A12生物活性的测定
1.4.1 抗菌活性 采用菌丝生长速率法[24]测定目标化合物A1~A12对6种供试植物病原真菌的体外抑菌活性。将已准确称量的5 mg待测化合物完全溶于体积分数5% DMSO水溶液中,以嘧菌酯原药和体积分数5%的DMSO水溶液作为空白对照;将10 mL待测液或对照溶液与90 mL无菌PDA培养基于50 ℃左右快速混匀,得到质量浓度为50 μg/mL的药液;趁热倒入已灭菌的培养皿内,每皿10 mL,冷却备用。将供试病原菌(菌饼直径5 mm)接种到上述培养皿中,每个测试组设3个平行[5],于湿度60%、28 ℃下恒温培养72 h,用十字交叉法测量菌落直径(mm),按下式计算菌丝生长抑制率(inhibition rate,IR):
IR=(dc-ds)/(dc-d0)×100%。
式中:dc为空白对照组菌落平均直径,ds为样品组菌落平均直径,d0为菌饼直径(5 mm),。
1.4.2 杀螨活性 用昆虫分离针挑取大小均一、活力较强的成年体兔痒螨作为受试对象[25]。分别称取待测样品A1~A12 5.0 mg,用0.1 mL DMSO超声溶解,加入0.1 mL吐温-80、0.8 mL质量分数0.9%的生理盐水,超声振荡下溶解均匀,配制得到浓度为5 mg/mL的供试药液。空白对照组CK为0.5 mL DMSO、0.5 mL的吐温-80和4.0 mL的质量分数0.9%生理盐水的混合液。在48孔细胞培养板的每孔中各加入0.2 mL供试药液和20只兔痒螨,每种药液设3组重复[26-28]。将培养板置于饱和湿度为22 ℃的培养箱内,24 h后检查结果,用昆虫分离针刺激所有不活动的兔痒螨,没有反应则表示死亡。以空白对照组CK为参照计算致死率:
致死率=死亡螨虫数/供试螨虫数×100%。
校正致死率=(药液组致死率-对照组致死率)/(1-对照组致死率)×100%。
2 结果与分析
2.1 目标化合物的基本性质
目标化合物A1~A12的理化性质及ESI-MS数据见表1,1H NMR(δ)及13C NMR(δ)数据见表2。
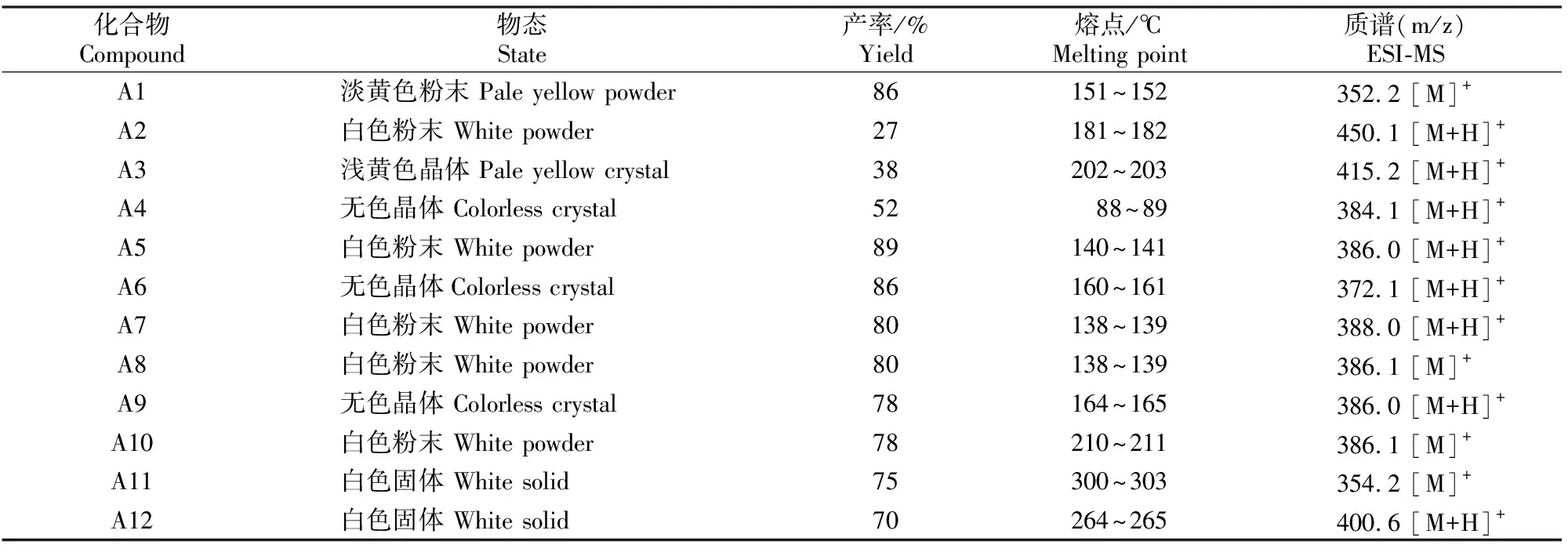
表1 12种化合物的理化性质及ESI-MS数据Table 1 Physico-chemical properties and ESI-MS data of the target 12 compounds

表2 12种目标化合物的1H NMR(δ)及13C NMR(δ)数据Table 2 1H NMR(δ) and 13C NMR(δ) data of the target 12 compounds

表2(续) Continued table 2
2.2 目标化合物的抗菌活性及构效分析
12种目标化合物(50 μg/mL)对6种植物病原菌的体外抑制活性测定结果见表3。表3显示,绝大多数化合物均表现出程度不一的抑菌活性;且对番茄早疫病菌的抑制率明显高于其他病原菌,但抑菌效果都不如药物对照嘧菌酯。对比发现,化合物A2、A6和A11对6种供试病原菌的抑制率均比原药别隐品碱有较大提高。这可能是因为,化合物A2是在原药别隐品碱的12-位碳引入卤族元素Br的产物,有效提高了化合物的抑菌活性;化合物A6是将原药的14-位羰基还原为羟基,化合物的抑菌活性也得到提高;化合物A11形成了哌啶环,增加了氢键配体,同时其空间位阻减小,化合物的抑菌活性得到提高。化合物A11对苹果腐烂病菌的抑菌活性最高,抑制率达73.28%,且其比化合物A6和A2对6种供试病原菌的体外抑制活性更具有广谱性。
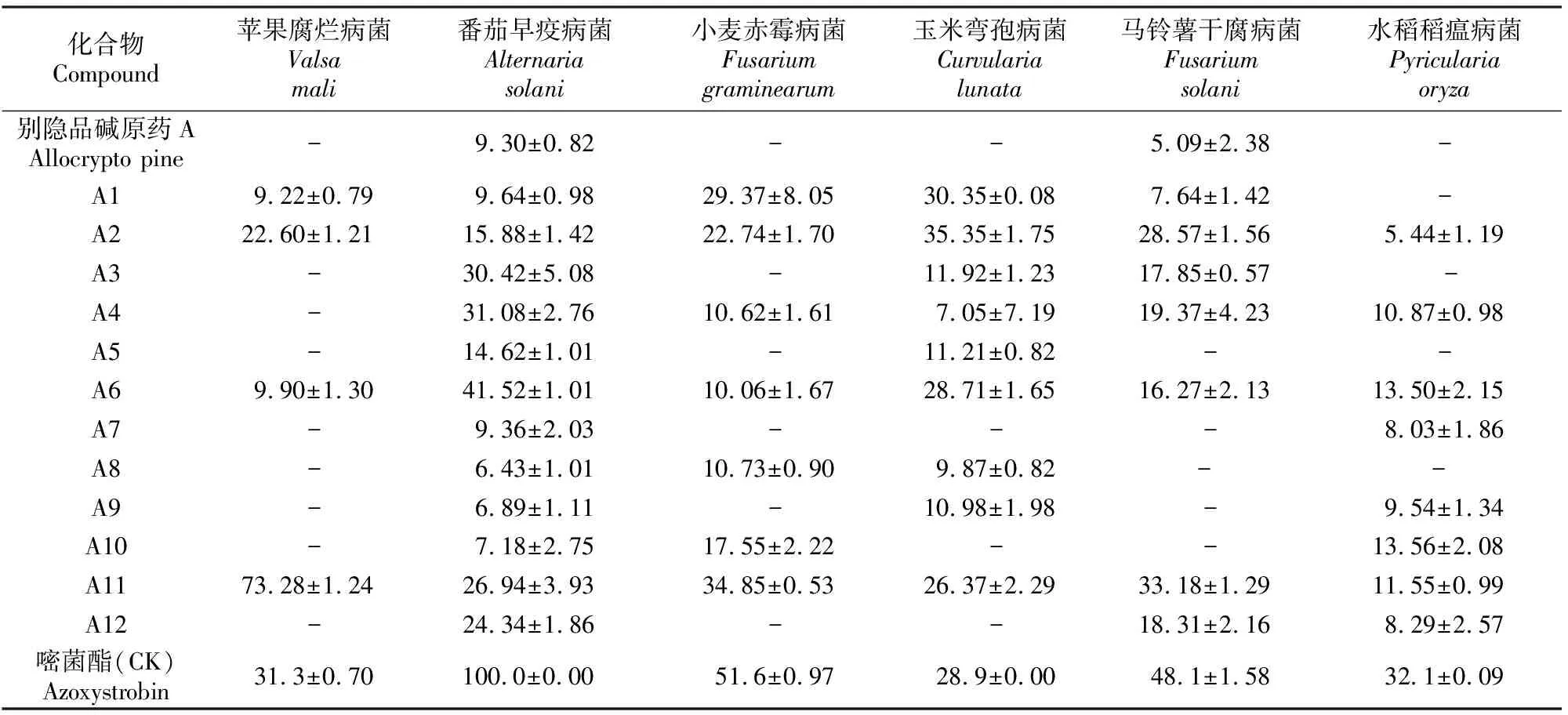
表3 目标化合物对6种植物病原菌的抑制活性Table 3 Antigungal activity of the target compounds against six plant pathogenic fungus %
注:表中“-”表示无活性。
Note:The “-” denotes that it is inactive.
2.3 目标化合物的杀螨活性
采用浸虫法测定别隐品碱原药及目标化合物的杀螨活性,结果见表4。由表4可以看出,在质量浓度为5 mg/mL时,别隐品碱及其修饰物大多都对兔痒螨有一定的杀灭活性。其中以化合物A11对兔痒螨的杀灭活性最好,致死率和校正致死率分别为61.55% 和58.2%。与原药相比,化合物A6对兔痒螨的致死率从30.42% 提高至40.30%,可见在别隐品碱14-位引入羟基有利于提高杀螨活性。
化合物A5比原药对兔痒螨的杀灭活性降低(表4),说明氧化7-位的氮原子不能提高化合物的杀螨活性。与化合物A6相比,化合物A7的杀螨活性相对较差,说明N被氧化的别隐品碱类化合物对螨虫的杀灭活性没有提高。与化合物A6相比,化合物A8的杀螨活性稍微降低,说明在N上多引入1个甲基不能提高此类化合物的杀螨活性。
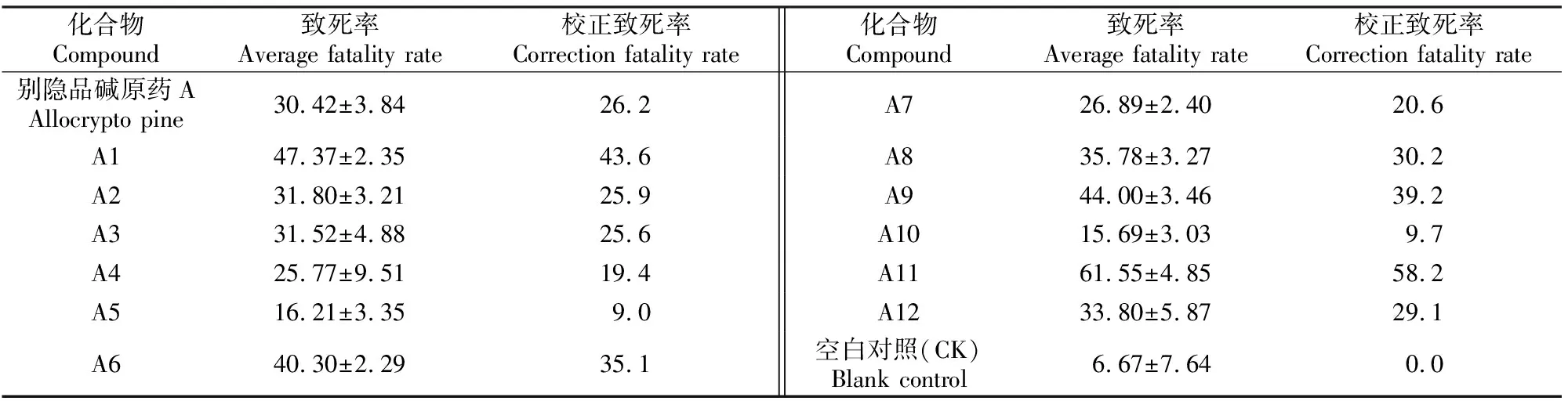
表4 目标化合物的杀螨活性 Table 4 Caricidal activity of the target compounds against Psoroptes cuniculi %
3 结 论
本研究发现,别隐品碱类修饰化合物大多表现出中低程度的杀菌和杀虫活性;其中化合物A1的抑制活性有较明显提高。与别隐品碱原药相比,化合物A2对苹果腐烂病菌、玉米弯孢病菌和马铃薯干腐病菌的抑菌活性都有所提高,可见苯环上的H被Br取代后,抑菌活性提高。在别隐品碱的14-位引入羟基有利于提高其杀螨活性,由此可知14-位羰基是别隐品碱的活性位点,将其还原成羟基后可以提高抗真菌活性和杀螨活性,还可拓宽其抗菌谱。
猜你喜欢
化学工程师(2022年3期)2022-04-19
云南农业科技(2022年1期)2022-01-27
上海化工(2021年2期)2021-04-23
今日农药(2017年7期)2017-08-09
中小学实验与装备(2016年1期)2016-04-19
科技与企业(2015年20期)2015-10-21
今日农药(2014年12期)2015-03-30
红领巾·探索(2014年8期)2014-10-10
中国农资(2012年31期)2012-08-15
浙江农业科学(2010年6期)2010-05-30
