
血清总同型半胱氨酸候选参考测量程序(液相色谱串联质谱法)的建立及性能评估
2018-11-29 08:06杨晓东邹继华邹炳德
检验医学 2018年11期
沈 敏, 杨晓东, 王 琳, 邹继华, 张 曼, 邹炳德
(1. 美康生物参考实验室,浙江 宁波 315104;2. 首都医科大学附属北京世纪坛医院检验科,北京 100038)
同型半胱氨酸(homocysteine,Hcy)是人体内蛋氨酸和半胱氨酸代谢的重要中间产物[1]。正常人体内Hcy浓度为5~15 μmol/L。由于遗传或者其他因素使人体内Hcy浓度持续高于正常值,即出现高同型半胱氨酸血症(hyperhomocysteinemia,HHcy)。近年来的研究结果表明,HHcy是心脑血管疾病的独立危险因素[2-3];同时,Hcy与冠状动脉疾病[4-5]、脑血管疾病[6-7]、糖尿病大血管病变[8]及静脉血栓形成[9-10]等多种疾病有着非常重要的关系。因此,开展Hcy普查对于某些疾病的预防、诊断及治疗具有非常重要的意义。血浆Hcy主要包括3种形式[11]:70%~80%为蛋白结合型Hcy,20%~30%为Hcy二聚物或混合Hcy,只有1%为游离型Hcy。临床上通常检测的是总Hcy。
目前,总Hcy的检测方法较多,包括循环酶法、荧光偏振法、酶联免疫法、高效液相色谱(high-performance liquid chromatography,HPLC)-电化学法、HPLC-荧光法、气相色谱质谱联用(gas chromatography-mass spectrometry,G C-M S)及液相色谱串联质谱(l i q u i d chromatography-tandem mass spectrometry,LCMS/MS)等[12-17]。在这些方法当中,生化法和免疫法虽然具有全自动、快速的优点,但其试剂昂贵,特异性较差,且检测结果的准确性和重复性不佳。HPLC、GC-MS虽然准确度较高,但样本需要进行衍生化等步骤处理,操作比较复杂且耗时。LC-MS/MS由于具有敏感性高、特异性好、准确度高等突出优点,近年来已被用于总Hcy的临床检测。
临床检验参考方法广泛应用同位素稀释质谱(isotope-dilution mass spectrometry,IDMS)原理,在检验医学溯源性联合委员会(the Joint Committee for Traceability in Laboratory Medicine,JCTLM) 2006年公布的国际临检参考方法列表中,有机小分子检验项目57种参考方法中有52种基于ID-MS原理[18]。总Hcy检测的参考方法也是基于ID-MS原理。目前,在JCTLM列表上公布的总Hcy参考方法有3种[19-20],均由美国国家标准与技术研究院(the National Institute of Standards and Technology,NIST)开发,对血清或血浆中总Hcy的检测结果比较理想。但由于其选用的标准物质为DL-高胱氨酸,同位素内标为氘8-高胱氨酸,二者分子结构均为2分子Hcy的巯基通过共价二硫键结合而成,需要对二者各自进行还原,才能进行准确定量,而该还原过程操作较为繁琐且耗时。因此,本研究在此基础上,直接以Hcy单体作为标准物质,以氘4-Hcy作为内标,进一步优化色谱和质谱检测条件,以期能基于LC-MS/MS技术建立一种简单、准确且稳定的候选参考测量程序,以实现血清总Hcy项目的量值传递,保证临床常规检测系统测量结果的准确性。
1 材料和方法
1.1 仪器与试剂
AB SCIEX QTRAP 5500液相色谱串联质谱系统(美国SCIEX公司)、SUPELCOSIL LC-CN 色谱柱(250 mm×4.6 mm,粒径5 μm,美国SUPELCO公司)、XS 205DU型分析天平(瑞士METTLER TOLEDO公司)、Heidolph Multi Reax漩涡混合器(德国Heidolph公司)、Eppendorf 5427R高速冷冻离心机(德国Eppendorf公司)、Eppendorf移液器(德国Eppendorf AG公司)、容量瓶(日本ASone公司)、Milli-Q超纯水系统(德国Millipore公司)。
DL-Hcy(即Hcy标准品)纯度为95.6%,购自美国Sigma公司;同位素内标DL-Hcy-2H4(简称Hcy-d4)购自美国IsoSciences公司,纯度为98.88%,同位素丰度为99.72%;正确度验证标准物质SRM 1950购自NIST;DL-二硫苏糖醇(薄层色谱级)和三氟乙酸(色谱级)购自美国Sigma公司;甲醇(Optima LC/MS级)、甲酸(Optima LC/MS级)、乙腈(Optima LC/MS级)均购于美国Thermo Fisher公司。实验用水采用Milli-Q超纯水系统制备。
1.2 方法
1.2.1 标准品与内标品制备 用重量法分别精密称取Hcy标准品、Hcy-d4内标,加水溶解并稀释混匀,分别制备出500 μmol/L Hcy标准储备液和100 μmol/L Hcy-d4标准储备液,避光2~8 ℃密封保存。精密称取不同质量的Hcy标准储备液,加入水稀释并混匀,分别制备出0.5、2.0、5.0、10.0、15.0、30.0、50.0、100.0、200.0 μmol/L共9个浓度梯度的Hcy标准工作液;同理,制备出10.0 μmol/L Hcy-d4标准工作液。以上溶液需计算溶液密度(g/mL),避光2~8 ℃密封保存。
1.2.2 样本前处理 用重量法移取不同浓度的Hcy标准工作液或血清样本100 μL 于1.5 mL离心管中,加入100 μL Hcy-d4标准工作液(重量法),然后再加入20 μL DL-二硫苏糖醇溶液,涡旋混合2 min后室温平衡15 min,加入200 μL乙腈[含0.05%三氟乙酸(V∶V)、0.1%甲酸(V∶V)]溶液,涡旋混合2 min后13 000×g离心5 min,取上清100 μL加入到进样瓶中,加入900 μL 10%甲醇(V∶V)水溶液稀释至1 mL,混匀,用于LC-MS/MS分析。
1.2.3 液相色谱条件 色谱柱为SUPELCOSIL LC-CN色谱柱,流动相A为0.1%甲酸甲醇溶液(V∶V),流动相B为0.1%甲酸水溶液(V∶V),流速为0.5 mL/min,柱温为30 ℃,进样量为3 μL,梯度洗脱(0→7 min,10%A;7→9 min,10%A→100%A;9→12 min,100%A;12→13 min,100%A→10%A;13→15 min,10%A)。
1.2.4 质谱分析条件 离子源为电喷雾电离源(electrospray ionization,ESI),离子化方式为正离子模式,离子化电压为5 500 V,雾化温度为550 ℃,碰撞气强度为Medium,气帘气、雾化气和辅助加热气分别为35 psi、55 psi、40 psi。以多反应监测(multiple reaction monitoring,MRM)模式扫描分析。具体参数见表1。
1.3 方法性能评估
参照美国临床实验室标准化协会(the Clinical and Laboratory Standards Institute,CLSI)C62-A文件[21]和C50-A文件[22]对建立的LC-MS/MS方法进行基本分析性能验证。

表1 质谱测定参数设置
1.3.1 标准曲线与线性评价 用重量法各取9个浓度梯度的Hcy标准工作液(0.5、2.0、5.0、10.0、15.0、30.0、50.0、100.0、200.0 μmol/L)100 μL,按照样本前处理条件进行相同处理,每个浓度重复检测2次,取均值。各浓度的检测偏移<15%, 且r>0.99可判断为呈线性。
1.3.2 检测限与定量限 用生理盐水对已知浓度的高浓度(10.91 nmol/g)血清样本进行稀释,制备包括预期的定量限浓度在内的3个浓度梯度的稀释样本,混匀,分别计算出每个浓度梯度样本的理论浓度(C1),然后将定量限样本各一式五份进行处理后检测,得到实际测定值(C2)。将同时满足偏移 <20%、变异系数(coefficient of variation,CV)<20%、信噪比(signal-to-noise ratio,S/N)≥1 0的浓度值定义为定量限,将S/N=3的计算浓度值定义为检测限。
1.3.3 基质效应 采用基质混合实验的方法来检测相对基质效应,即通过检测血清基质样本、溶液基质标准溶液、血清和溶液1∶1(质量比)混合物、血清和溶液80∶20(质量比)混合物、血清和溶液20∶80(质量比)混合物这5种基质样本(各5份)中标准与内标的峰面积比值来计算相对基质效应。第1组:100 μL标准工作液+100 μL内标,共5份样本,经前处理进样得到标准与内标峰面积比值的平均值A;第2组:100 μL血清+100 μL内标,共5份样本,经前处理进样得到标准与内标峰面积比值的平均值B;第3组:50 μL血清+50 μL标准工作液+100 μL内标,共5份样本,经前处理进样得到标准与内标峰面积比值的平均值C;第4组:80 μL血清+20 μL标准工作液+100 μL内标,共5份样本,经前处理进样得到标准与内标峰面积比值的平均值D;第5组:20 μL血清+80 μL标准工作液+100 μL内标,共5份样本,经前处理进样得到标准与内标峰面积比值的平均值E。以上操作均用重量法。1∶1混合物相对基质效应 =[C-(A+B)/2]/[(A+B)/2]×100%;80∶20混合物的相对基质效应=[D-(0.2A+0.8B)]/(0.2A+0.8B)×100%;20∶80混合物的相对基质效应=[E-(0.8A+0.2B)]/(0.8A+0.2B)×100%。如果3种混合物中标准与内标的峰面积比值与对应比例血清基质样本和溶液基质标准溶液中标准与内标的峰面积比值的均值差异 <20%,则视为无相对基质效应。
1.3.4 携带污染 在评估携带污染时,先重复进样低值样本(定量限浓度样本),然后交替进样高值(79.56 nmol/g)样本(H)和低值0.31 nmol/g样本(L),进样顺序如下:L1、L2、L3、H1、H2、L4、H3、H4、L5、L6、L7、L8、H5、H6、L9、H7、H8、L10、H9、H10、L11。比较第1次进样(L1)后跟随的低值样本检测结果的平均值A(即L2、L3、L6、L7、L8的平均值)与随后进样的高值样本后跟随的低值样本检测结果的平均值B(即L4、L5、L9、L10、L11的平均值)的差异,若低于预定标准[以20%为限,携带污染率=(B-A)/A×100%],则认为在测定该高值及以下浓度时不存在明显的携带污染。
1.3.5 精密度评价 采用低(11.29 μmol/L)、中(48.83 μmol/L)、高(81.86 μmol/L)3个浓度的混合血清作为待测样本,将混合血清分装后-70 ℃保存,对每个浓度样本每批次各5份进行样本前处理,连续测定3个批次,分别计算批内CV和批间CV。
1.3.6 正确度验证 分别采用加标回收和测定有证参考物质2种方式对方法的正确度进行评价。(1)加标回收实验:取3个已知浓度(10.91、47.60、79.56 nmol/g)的混合血清,采用重量法各取0.5 mL,加入50.58 μmol/L Hcy标准工作液0.5 mL(重量法),混匀,制备出3个浓度的加标样本,分别计算出理论浓度,然后将各个浓度的加标样本(各5份)进行处理后进样检测,得到实际测定值,计算加标回收率。回收率在95%~105%之间为可接受。(2)有证参考物质测定:采用本研究建立的LC-MS/MS方法测定NIST SRM 1950,对每个批次5份样本进行前处理,连续测定2个批次,考察方法的正确度。
1.3.7 稳定性 目标分析物的稳定性对检测结果的影响非常大,必须对其加以评估。以精密度验证样本中的低浓度血清样本为稳定性研究样本,通过以下2个方面评估目标分析物的稳定性:(1)在生物基质中的稳定性,包括室温放置4~24 h的稳定性、2~8 ℃保存1~4 d的稳定性及反复冻融的稳定性;(2)处理后样本的稳定性,包括处理后室温放置4~24 h的稳定性、处理后自动进样器放置4~24 h的稳定性。
1.4 不确定度评估
依据《测量不确定度表示指南》(Guide to the Expression of Uncertainty in Measurement,简称GUM),测量结果的浓度计算公式为C=(A/AIS-b)/k×fc×fm,式中C为总Hcy的测量浓度(μmol/L)、A/AIS为样本与内标的峰面积比值、b为标准曲线截距、k为标准曲线斜率、fc为标准与内标溶液浓度修正因子、fm为样本处理修正因子。从标准工作液配制、样本处理、仪器检测及重复测量4个方面分析不确定度来源,并量化各个分量。
2 结果
2.1 标准曲线与线性评价
LC-MS/MS检测血清总Hcy的色谱图见图1,标准曲线见图2,线性范围为0.5~200.0 μmol/L。
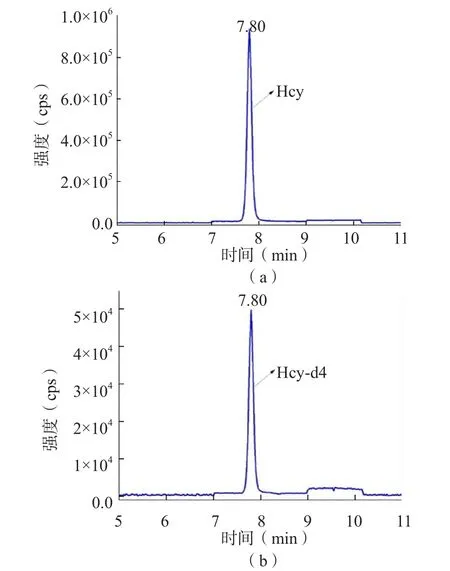
图1 Hcy和Hcy-d4的色谱图
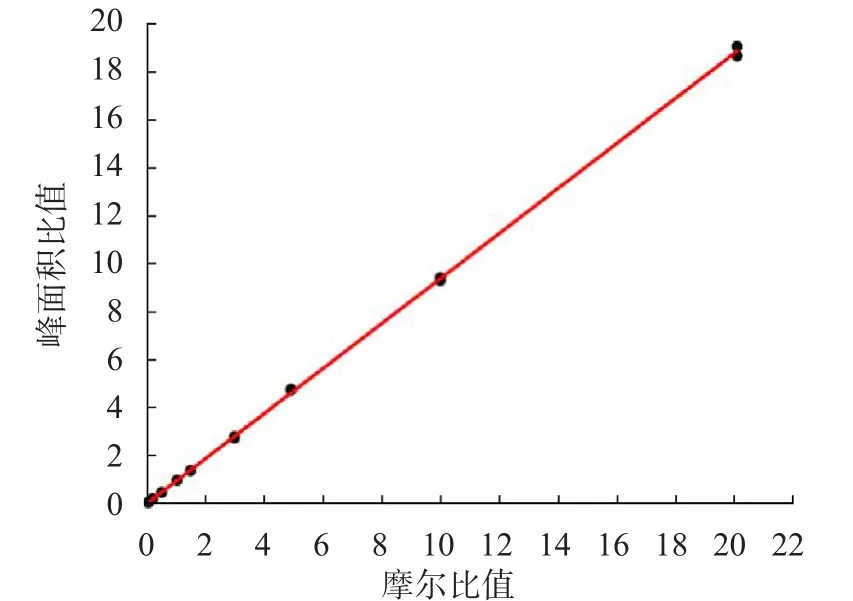
图2 LC-MS/MS检测血清总Hcy的标准曲线
2.2 检测限与定量限
LC-MS/MS检测血清总Hcy的定量限为0.31 nmol/g,检测限为0.06 nmol/g。见表2。
2.3 基质效应
采用基质混合实验考察相对基质效应。5份溶液基质样本的峰面积比值分别为0.907、0.920、0.920、0.920、0.920,平均值为0.917。5份血清基质样本的峰面积比值分别为1.030、1.033、1.047、1.040、1.033,平均值为1.037。5份1∶1混合物样本的峰面积比值分别为0.970、0.953、0.957、0.957、0.953,平均值为0.958。5份80∶20混合物样本的峰面积比值分别为0.993、0.998、0.987、1.003、0.995,平均值为0.995。5份20∶80混合物样本的峰面积比值分别为0.924、0.925、0.919、0.923、0.922,平均值为0.923。计算得到相对基质效应分别为1.94%、1.91%、1.78%。
2.4 携带污染
通过多次高值样本和低值样本交替重复进样,计算得携带污染率为2.86%,因此可认为该方法不存在明显的携带污染。
2.5 精密度评价
采用LC-MS/MS定量检测混合血清总Hcy浓度,批内、批间CV分别为<2%和<1%,见表3。
2.6 正确度验证
2.6.1 加标回收实验 加标后3种浓度的血清样本平均回收率分别为99.8%、100.2%、100.8%,准确度符合要求,见表4。
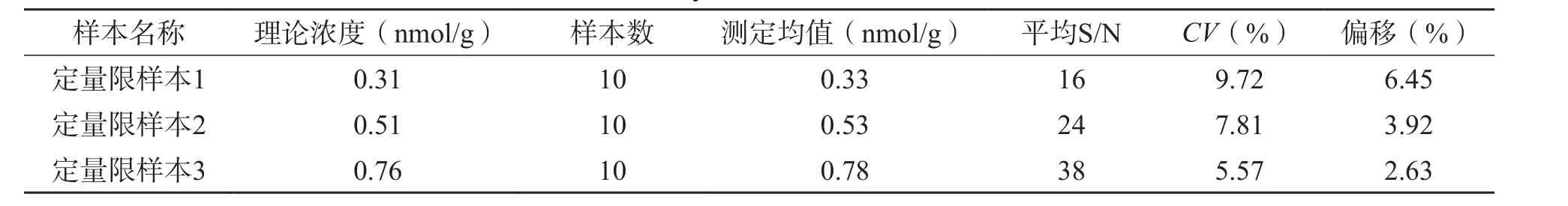
表2 Hcy的定量限评估结果
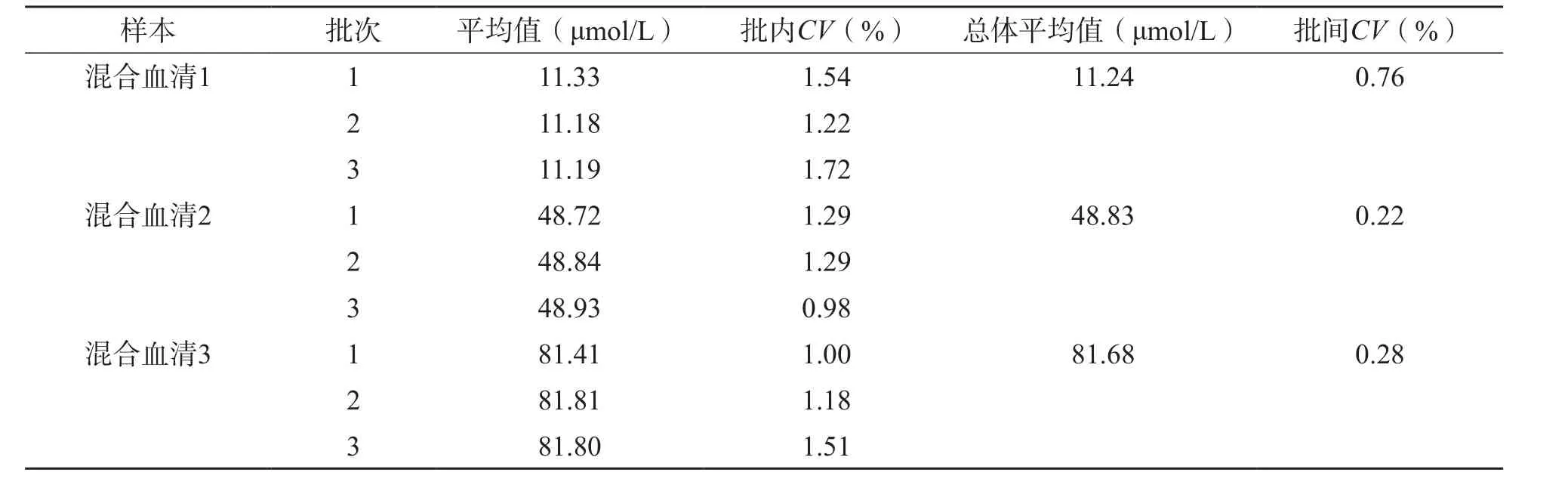
表3 LC-MS/MS精密度评价结果
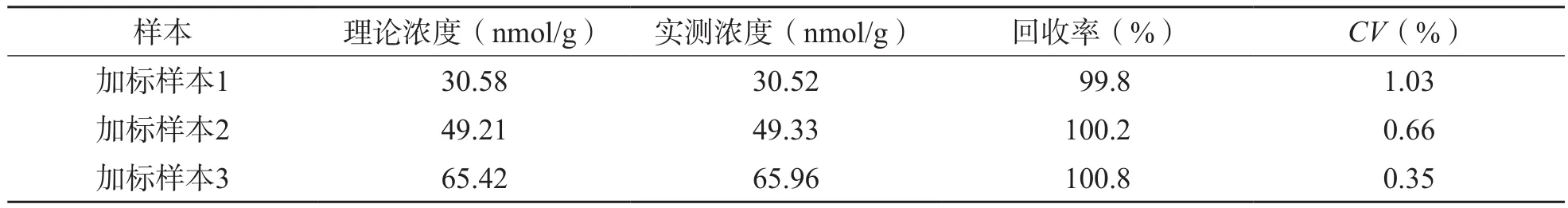
表4 加标回收率测定结果
2.6.2 参考物质测定 采用NIST SRM 1950标准物质[证书浓度为(8.50±0.20)μmol/L]进行正确度验证,连续测定2个批次,其测定均值、CV和偏移分别为8.55 μmol/L、1.65%、0.59%和8.49 μmol/L、1.35%、-0.12%,具有很高的正确度。
2.7 稳定性
2.7.1 在生物基质中的稳定性 低浓度血清样本在室温[(23±2 )℃]放置24 h、2~8 ℃放置4 d、反复冻融5次的情况下稳定。
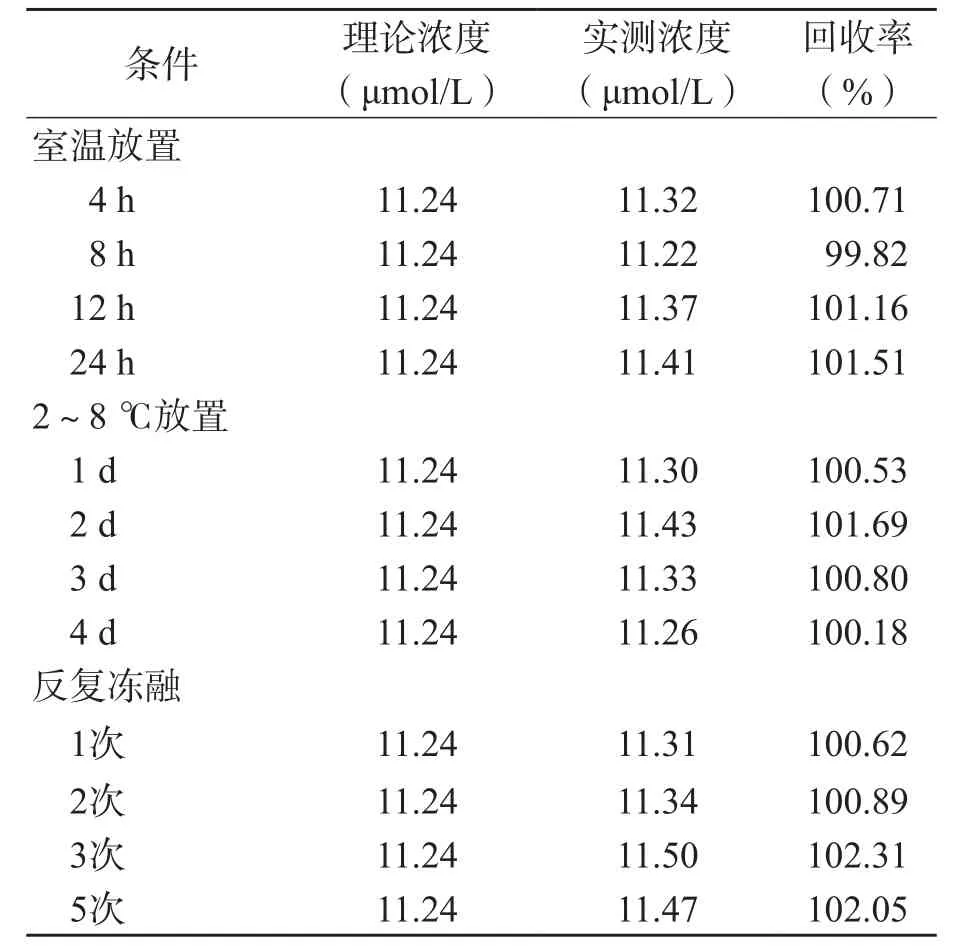
表5 生物基质样本中Hcy的稳定性
2.7.2 处理后样本的稳定性 样本处理后室温[(23±2 )℃]放置24 h和自动进样器(温度为10 ℃)放置24 h均非常稳定。
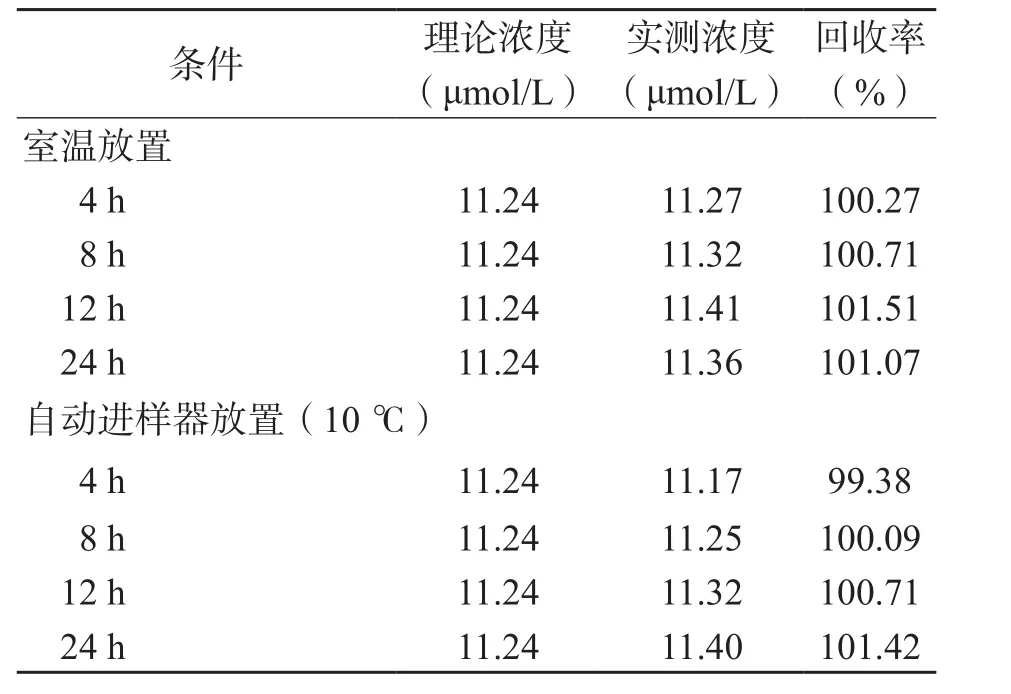
表6 处理后样本中Hcy的稳定性
2.8 不确定度评估
从标准工作液配制、样本处理、仪器检测及重复测量4个方面评估精密度实验测定的3种浓度样本测量结果的不确定度。结果显示候选参考测量程序测定血清总Hcy浓度产生的不确定度较为合理。见表7。

表7 3种不同浓度样本测定结果不确定度的评估结果
3 讨论
HHcy是心脑血管疾病的独立危险因子。体内Hcy浓度升高与神经系统疾病、高血压、糖尿病、肾病、孕妇流产、新生儿缺陷等多种疾病有关。目前,临床上已经将Hcy列为多种疾病诊断的重要指标。快速、准确地检测体内Hcy浓度可以为某些疾病的预防、诊断及治疗提供依据。因此,开发出快速、精准的检测方法显得尤为重要。LC-MS/MS结合了液相色谱分离与质谱质量分离的优点,敏感性高、特异性好。基于同位素稀释的LC-MS/MS保证了检测结果的准确度和精密度,近年来已被用于总Hcy的临床检测。
本研究基于JCTLM公布的总Hcy参考测量程序,建立一种简单、准确且稳定的候选LC-MS/MS参考测量程序。在标准物质和同位素内标的选择上,NIST选用的标准物质为DL-高胱氨酸,同位素内标为氘8-高胱氨酸,而本研究选择Hcy单体作为标准物质,氘4-Hcy作为同位素内标,无需还原步骤,直接以此建立标准曲线进行定量。LC-MS/MS检测总Hcy的过程涉及3个关键步骤:样本前处理、色谱条件和质谱条件的选择。在样本的前处理上,本研究选择以二硫苏糖醇为还原剂,在NIST参考方法的基础上对其浓度和加入量进行了微调,以保证所有结合型Hcy全部还原为游离型;同时,延长了蛋白沉淀后的离心时间,在保证检测敏感性的前提下对上清液进行了稀释,以减小基质效应的影响。在色谱条件的选择上,本研究选用的色谱柱、流动相、柱温、流速与NIST参考方法保持一致,但对其梯度洗脱程序进行了优化,降低了流动相中水相的初始比例,同时减缓了梯度,以减小各种内源性干扰物质对目标分析物的影响。质谱条件的优化主要是调整各个参数,使Hcy分子的母离子得到最佳的响应值。本研究使用ESI对Hcy进行Q1全扫描,获得m/z 136.1的[M+H]+峰,内标的[M+H]+峰为m/z 140.1,以各自的分子离子峰为母离子,对离子源参数进行优化,使其各自的响应值达到最佳,然后对其各自的子离子进行全扫描,确定特征碎片离子,组成MRM离子通道。本研究Hcy定量检测的MRM离子通道为m/z 136.1/90.0,内标检测定量的通道为m/z 140.1/94.0。
参照CLSI C62-A文件和C50-A文件对建立的候选参考方法进行分析性能验证。在建立方法时,所有的溶液配制和样本移取均采用重量法;在进行标准曲线拟合时,以标准品定量离子峰面积与内标峰面积的比值为纵坐标,以标准品与内标的摩尔比为横坐标,用最小二乘法进行直线拟合。结果显示LC-MS/MS检测总Hcy的线性范围为0.5~200.0 μmol/L,定量限和检测限分别为0.31和0.06 nmol/g。该方法的敏感性和线性范围完全能够满足常规检测的溯源要求。相对基质效应为2.04%,证明采用同位素内标能够校正基质效应的影响。精密度评价结果显示,本方法的批内、批间CV分别为<2%和 <1%,3种浓度的加标样本平均加标回收率分别为99.8%、100.2%、100.8%。Hcy的标准物质SRM 1955已经从NIST标准物质列表中删除,本方法测定NIST SRM 1950标准物质的偏移<1%,精密度和正确度符合参考测量程序的要求。LCMS/MS参考测量程序的定量分析方法一般采用包括法。标准溶液中被测组分的量与对应内标的量的比值控制在0.9和1.1,样本中被测组分的量与内标的比值尽量控制在1.0左右。该方法能非常准确地测定待测组分的浓度,但操作较繁琐,需要对待测样本先测定一个初步浓度值,再采用包括法进行准确定量。最近,JCTLM公布了一种采用标准曲线法对25-羟基维生素D进行准确定量的参考测量程序[23],其采用高、低浓度2条标准曲线,对高浓度样本采用高浓度标准曲线进行定量,对低浓度样本采用低浓度标准曲线进行定量,避免了二次测量,从而简化了操作。本研究采用标准曲线法建立了血清Hcy的参考测量程序,其正确度和精密度符合参考测量程序的要求,表明标准曲线法也可以作为准确定量的方法。
综上所述,本研究建立的基于LC-MS/MS的血清总Hcy候选参考测量程序具有操作简便、特异性强、准确性好、运行稳定等特点,能够用于总Hcy项目的量值溯源和标准化及常规检测系统的正确度评价,在临床检验中具有实用价值。
猜你喜欢
中国氯碱(2022年10期)2022-11-22
口腔护理用品工业(2021年4期)2021-11-02
化学工程师(2020年7期)2020-09-07
世界科学技术-中医药现代化(2020年2期)2020-07-25
中成药(2018年12期)2018-12-29
中成药(2018年6期)2018-07-11
制造技术与机床(2017年9期)2017-11-27
中成药(2017年6期)2017-06-13
中国粮油学报(2016年5期)2016-01-23
中国氯碱(2015年5期)2015-06-15
