
黑曲霉高效敲除体系构建及tpsA基因的敲除
2018-11-09 05:37刘玲张鸿飞张岚秦郦王德培
食品研究与开发 2018年22期
刘玲,张鸿飞,张岚,秦郦,王德培,3,*
(1.工业发酵微生物教育部重点实验室,天津300457;2.省部共建食品营养与安全国家重点实验室,天津300457;3.天津科技大学生物工程学院,天津300457)
黑曲霉(Aspergillus niger)作为一种重要的工业生产安全菌株之一,可用于有机酸、淀粉酶、酸性蛋白酶等的工业生产中。同时也被广泛应用于医药,饲料生产,食品加工,废料处理等领域[1]。因此进一步从分子方面深入研究工业菌株黑曲霉的代谢机理,可以从微观的角度调控基因的表达,为获得高产工业黑曲霉菌株奠定了理论基础。目前,基因敲除是研究基因功能最为便捷准确的分子生物学改造手段之一,主要应用于对已知DNA序列的基因功能和基因调控机理研究。基因敲除以基因同源重组技术为基础,通过构建带有同源重组的DNA片段的基因敲除载体,将其转化入目的细胞中,部分或者完全取代原始基因组中的等位基因,通过对突变体表型的分析从而反向研究基因功能的方法[2]。
工业黑曲霉基因敲除体系依赖于根癌农杆菌介导的黑曲霉转化体系(Agrobacterium tumefaciens-mediated transformation,ATMT),该转化体系的条件优化已经完成,大大减少了丝状真菌的转化操作,提高了载体上的同源DNA片段的插入效率[3]。农杆菌介导转化法虽然能大幅度地提高同源重组片段的转化效率,但是由于T-DNA自身会随机整合到基因组中,携带同样的抗性标记造成假阳性率高,从而使筛选同源双交换产生的基因敲除阳性转化子工作的难度增大。另一方面,等位基因同源重组的效率相对T-DNA随机插入的效率低很多,造成基因敲除阳性转化子的数量远远少于随机插入的转化子的数量,因此需要筛选大量转化子才能得到正确的基因敲除转化子。单纯疱疹病毒胸苷激酶(herpes simplex virus thymidine kinase,HSVtk)可在丝状真菌中表达,将培养基中的特定化合物5-氟脱氧尿苷(5-fluoro-2'-deoxyuridine,F2dU)转变为真菌的毒性物质,导致真菌致死[4],因此,可在TDNA的同源重组DNA片段上游插入致死基因HSVtk基因序列,从而减少黑曲霉假阳性转化子,提高正确基因敲除菌株的比例,从而提高筛选效率。
海藻糖是由两个葡萄糖分子以1,1-糖苷键构成的非还原性二糖,其代谢的前体物质为6-磷酸海藻糖。6-磷酸海藻糖主要是由尿苷二磷酸葡萄糖和6-磷酸葡萄糖经海藻糖6-磷酸合成酶催化合成6-磷酸海藻糖。6-磷酸海藻糖是6-磷酸葡萄糖分支代谢流的重要物质,显著影响6-磷酸葡萄糖合成柠檬酸的转化效率,因此敲除海藻糖-6-磷酸合成酶(trehalose-6-phosphate synthase,tpsA),可以调控碳源在主代谢流的积累量,从而提高黑曲霉的柠檬酸产量。本文首先构建具有致死基因HSVtk的工程菌株,获得致死筛选浓度为10 μmol/L 5-氟脱氧尿苷,然后构建具有单纯疱疹病毒胸苷激酶和潮霉素双标记以敲除tpsA质粒,通过反向筛选,减少假阳性率,高效获得tpsA同源重组敲除菌株。
1 材料与方法
1.1 材料
1.1.1 菌株,质粒和引物
试验所用菌株和质粒见表1。试验中用到的引物见表2。

表1 试验所用的菌株和质粒Table 1 Strains and plasmids used in test

表2 试验中用到引物Table 2 The primers used in test
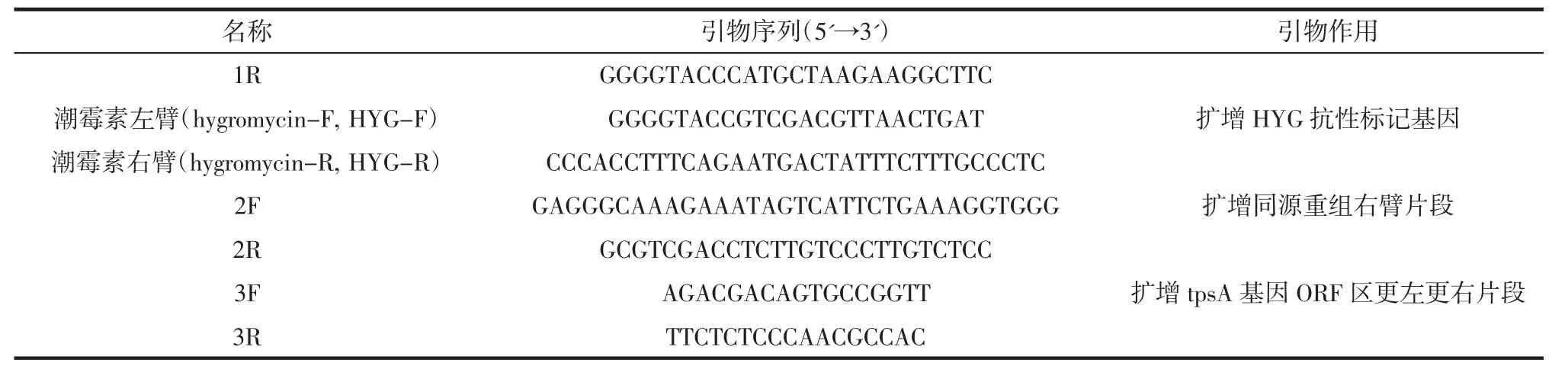
续表2 试验中用到引物Continue table 2 The primers used in test
1.1.2 培养基
肉汤(Luria-Bertani,LB)培养基用于大肠杆菌和农杆菌液体培养,完全培养基(complete medium,CM)和马铃薯葡萄糖琼脂(potato dextrose agar,PDA)培养基用于黑曲霉的培养,配制方法参照文献[5]。诱导培养基(induction medium,IM),农杆菌侵染所需培养基及试剂参考文献[6]配制。
1.2 试剂与溶液的配制
Taq DNA 聚合酶(5 U/μL)、Prime STAR HS DNA聚合酶(2.5 U/μL)、RNaseA(5 mg/mL)、T4 DNA 连接酶(10 U/μL)、DL5000 DNA Maker、DL10000 DNA Maker、限制性内切酶 EcoRI(15 U/μL)、SalI(15 U/μL)、KpnI(10 U/μL):TaKaRa公司;5-氟脱氧尿苷、潮霉素 B、乙酰丁香酮(acetosyringone,AS):Solarbio公司;其他试剂:天津市北方天医化学试剂厂。
潮霉素B(hygromycin):潮霉素B 100 mg,ddH2O定容至1 mL。母液浓度为100 mg/mL,工作浓度为200 μg/mL。
1.3 根癌农杆菌介导的黑曲霉转化体系
将实验室保存的质粒p80-HSVtk通过电转化的方法,导入根癌农杆菌Agrobacterium tumefaciens(AGL1)中,再利用农杆菌AGL1介导侵染黑曲霉,完成黑曲霉的遗传转化,具体操作方法参见曹张磊等[3]的方法进行。
1.4 黑曲霉对F2dU的敏感性实验
为确定黑曲霉是否适用于5-氟脱氧尿苷参与下的反向筛选体系,进行黑曲霉对F2dU的敏感性实验。将质粒p80-HSVtk电转化进农杆菌AGL1中,使用携带质粒p80-HSVtk的农杆菌介导侵染原始菌株黑曲霉10142,从而获得一株含有致死基因HSVtk的随机插入黑曲霉转化子7-1。取200 μL浓度均为1.3×106个/mL的突变株7-1和原始菌株的孢子悬液分别涂布于 F2dU 终浓度为 0、10、25、50、150、300 μmol/L的CM培养基中,35℃静置培养5 d,观察结果。
1.5 敲除质粒p81的构建
海藻糖-6-磷酸合成酶(tpsA)基因的同源重组敲除片段的构建使用了融合PCR的方法,将同源重组左右臂,潮霉素抗性标记以及致死基因用常规PCR分别扩增后,融合成为tpsA基因敲除框,酶切连接完成p81质粒的构建。首先以p44质粒为模板,以HYG-F/HYG-R为引物(本文所用引物碱基序列见表2),常规PCR反应扩增1.4 kb的潮霉素抗性片段。同时以pBHt2-tk质粒为模板,以HSVtk-F/HSVtk-R为上下游引物,PCR扩增1.8 kb的HSVtk基因片段。以黑曲霉基因组为模板,以1F/1R和2F/2R为引物,PCR扩增长度均为1 kb左右的tpsA基因同源重组左右臂片段。其中引物1F的5’端含有与HSVtk-R互为反向互补的接头序列,引物2F的5’端含有与HYG-R互为反向互补的接头序列。从而获得末端含有相应互为反向互补序列的4个PCR产物。使用融合PCR的方法扩增2.8 kb的HSVtk和同源左臂的融合片段,使用限制性内切酶KpnI和EcoRI双酶切连接的方法将此片段连接到质粒p40中。同样的方法,使用限制性内切酶KpnI和SalI将HYG和同源右臂的融合片段双酶切连接到过程质粒p40-HSVtk-L中。从而完成敲除质粒p81的构建。
1.6 tpsA基因敲除阳性菌株的筛选验证
将农杆菌AGL1介导侵染黑曲霉后的孢子,涂布于浓度为200 mg/mL的潮霉素B的抗性平板上,待长出单菌落点接到10 μmol/L浓度的F2dU平板中,35℃培养5天后,挑出能正常生长菌株即可能的同源重组阳性转化子,提取基因组并利用PCR的方法进行分子生物学验证。
2 结果与讨论
2.1 F2dU的筛选浓度的确定
为了确定F2dU的筛选浓度,必须获得一株含有HSVtk基因的黑曲霉随机插入转化子。将质粒p80-HSVtk电转化进农杆菌AGL1中,使用携带质粒p80-HSVtk的农杆菌介导侵染原始菌株黑曲霉10142,通过潮霉素抗性筛选获得一株含有致死基因HSVtk的随机插入黑曲霉转化子7-1。在F2dU终浓度分别为0、10、25、50、150、300 μmol/L 的 CM 平板上,观察突变株7-1和原始菌株对F2dU的敏感性。观察结果如图1所示。
含有HSVtk基因的黑曲霉转化子在浓度为10 μmol/L的F2dU的CM平板上,孢子完全无法萌发。因此选用10 μmol/L的F2dU当作转化子的筛选浓度即可抑制含HSVtk的T-DNA随机插入转化子的生长,从而大大减少筛选操作,提高阳性转化子的比例。同时原始菌株在含有10 μmol/L F2dU的CM平板上生长并不受到影响。证明该反向筛选方法可减少随机插入转化子的概率,提高黑曲霉原位同源重组转化子的筛选效率。

图1 黑曲霉7-1和原始菌株在含有不同浓度F2dU的CM培养基上培养3 d的生长情况Fig.1 The growth of 7-1 and A.niger 10142 on CM plates with the different F2dU concentrations after 3 d
2.2 tpsA基因敲除质粒的成功构建
通过常规PCR反应,得到长度分别为1.8、1.0、1.4、1.0 kb的HSVtk基因片段,同源重组左臂片段,HYG基因片段和同源重组右臂片段。利用融合PCR,将HSVtk基因和同源左臂这2个DNA片段末端反向互补序列相互结合,构建2.8 kb的HSVtk和同源左臂的融合片段,同样的方法,得到2.4 kb的HYG和同源右臂的融合片段。先通过KpnI和EcoRI处理质粒p40和2.8 kb的融合片段,再过夜连接,转化大肠杆菌DH5α感受态细胞,筛选阳性转化子后得到过程质粒p40-HSVtk-L。再使用同样的方法,使用KpnI和SalI将2.4 kb的融合片段连接到过程质粒p40-HSVtk-L中,完成敲除质粒p81的构建,见图2。

图2 tpsA基因敲除质粒p81构建Fig.2 The construction of plasmid p81
2.3 tpsA基因敲除菌株的构建
将构建好的p81tpsA基因敲除质粒,转化入大肠杆菌感受态细胞中。大肠杆菌菌落PCR验证正确后提取质粒,具体质粒提取方法参见天根质粒回收试剂盒说明书。接着将质粒电转入农杆菌感受态细胞中,菌落PCR验证正确后,准备农杆菌侵染黑曲霉实验。通过根癌农杆菌介导的黑曲霉转化实验,完成黑曲霉的遗传转化。挑选在10 μmol/L F2dU的CM平板上仍能正常生长的16个tpsA基因敲除转化子,提取转化子基因组后对其进行分子生物学验证。琼脂糖凝胶电泳结果如图3(a)所示。以质粒p81为阳性对照,以原始基因组为阴性对照。使用引物1F/2R分别能扩增出3.5 kb和2.8 kb的DNA条带。以转化子的基因组为模板,同样使用 1F/2R 引物,其中(2、4、5、6、7、10、11、13、14、16号泳道)转化子能扩增出3.5 kb的单一条带,没有原始基因组的2.8 kb的条带,说明这10个转化子的tpsA基因编码框内的700 bp已经被1.4 kb的潮霉素抗性片段同源重组双交换所替代,从而破坏tpsA基因,为正确的同源重组双交换敲除菌株。其余6个转化子中,有4个泳道的转化子琼脂糖凝胶电泳图显示为2.8 kb包含原始菌株tpsA基因的ORF区和3.5 kb的T-DNA中随机插入的敲除框片段的双电泳条带,为T-DNA随机插入的黑曲霉转化子;剩下两个为原始基因的2.8 kb的单一条带。统计数据,tpsA基因敲除转化子在总转化子中比例为63%。PCR初步验证正确的转化子,对其进行左臂更左片段和右臂更右片段的扩增。使用3F/HYG-R引物,以转化子的基因组为模板,对其进行左臂更左片段的扩增,如图3(b)所示。扩增出了与预期结果相符的3 000 bp的片段。同样使用HYG-F/3R引物,对转化子进行右臂更右片段的扩增,结果如图3(c)所示。结合图 3(b)和图 3(c)的琼脂糖凝胶电泳图的结果,说明转化子均为原位同源重组双交换的敲除菌株。成功构建tpsA基因的敲除菌株Aspergillus niger101-ZM19,基因敲除流程图如图4所示。

图3 黑曲霉tpsA基因敲除转化子的分子生物学验证Fig.3 Verification of A.niger transformants

图4 基因敲除流程图Fig.4 The schematic of tpsA gene replacement in A.niger
2.4 摇瓶发酵验证敲除突变株的有机酸产量
海藻糖合成酶中的tpsA与海藻糖合成有关,tpsA通过抑制己糖激酶的活性来控制葡萄糖流向糖酵解通路。葡萄糖进入细胞以后,首先经己糖激酶催化,生成6-磷酸葡萄糖。tpsA基因被敲除时,己糖激酶的活性不再受抑制,磷酸化的己糖将大量积累,促进葡萄糖大量流向糖酵解途径和三羧酸循环,从而有利于菌体代谢产酸,提高黑曲霉敲除突变株的有机酸产量。
使用带渣玉米液化液进行黑曲霉的摇瓶发酵实验,控制初始还原糖含量在16%,500 mL三角瓶的玉米液化液的装液量为50 mL,突变株和原始黑曲霉孢子接种量均为105个/mL,280 r/min,35℃发酵72小时后测发酵液中的有机酸含量。使用酸碱滴定的方法,测得突变株的有机酸含量比原始菌株提高了6.8%,产酸结果如图5所示。
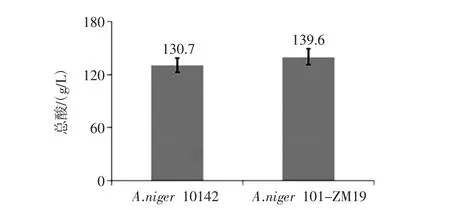
图5 黑曲霉转化子72 h摇床产酸验证Fig.5 The shaking acid of Aspergillus niger transformants
3 结论与讨论
黑曲霉作为工业生产有机酸和酶制剂的重要菌株,其基因功能研究对于未来菌株的分子生物学改造具有指导性意义。基因敲除是研究基因功能最直接、有效的技术手段。黑曲霉作为真核生物,其基因敲除转化子的筛选难度较大,正确突变株的概率较低,因此一个高效黑曲霉基因敲除体系的成功构建会加快黑曲霉基因工程改造的速度[7]。本文利用HSVtk基因在黑曲霉中表达,导致其具有F2dU培养物中致死的特性,构建在同源重组上游序列T-DNA插入HSVtk基因,使非同源重组转化子在含F2dU培养基中致死,从而大大减少假阳性转化子的数量,提高同源重组敲除突变株的筛选效率。
文献报道可知,稻瘟病菌和尖孢镰刀菌对F2dU不敏感,当HSVtk基因的随机插入基因组后,在含有浓度为0.5 μmol/L的F2dU培养基中即可导致稻瘟病菌和尖孢镰刀菌的随机插入转化子致死[8]。本试验发现黑曲霉CGMCC10142菌株随机插入HSVtk基因,其转化子在F2dU终浓度为10 μmol/L时,表现出致死特征。表明不同丝状真菌对外源基因的表达存在较大差异,因此,不同真菌随机插入HSVtk基因转化子对F2dU的敏感性不同,必须进行目标菌株对F2dU的敏感性试验,以及转化HSVtk基因后对F2dU的敏感浓度。
本试验黑曲霉CGMCC10142孢子对F2dU的不敏感性,在300 μmol/L的F2dU的筛选浓度下仍有50%以上的萌发率,随机插入HSVtk基因转化子7-1显著提高了F2dU的敏感性,致死浓度降低到10 μmol/L,且致死率为100%。因此可以将HSVtk基因连接于敲除框的上游或下游,不易发生缺失的T-DNA区段,从而获得黑曲霉的高效基因敲除体系。本文通过应用该体系对黑曲霉tpsA基因的敲除,正确的基因敲除突变株在总转化子中的比例达到63%,大大提高了阳性转化子的筛选概率,表明该体系可高效敲除黑曲霉基因。
对于转化子的验证,本试验除了验证敲除框的完整性,也验证了敲除框同源重组的位置。在同源双交换的左臂更左70 bp和右臂更右80 bp分别设计引物进行验证,从而将PCR验证转化子的正确性提高到100%,得到一株tpsA基因敲除突变株A.niger 101-ZM19。对其进行生长形态观察及碳源利用效率的研究,发现其特性与出发菌株基本保持一致,没有明显变化。有研究者发现,在植物中tpsA基因主要控制海藻糖的合成,从而提高植物的抗逆性能[8-9]。而在丝状真菌中实现tpsA基因敲除后,其分生孢子的耐热性能降低,但其生长特性及对葡萄糖的利用速率几乎没有改变[10]。同时,黑曲霉tpsA基因敲除菌的柠檬酸积累时间会提前[11]。因此对于黑曲霉tpsA基因的敲除菌A.niger 101-ZM19的产酸性能进行研究,发现其产酸性能得到提高,相对于出发菌株的柠檬酸产量提高了6.8%。
猜你喜欢
汉字汉语研究(2021年2期)2021-08-30
江西农业学报(2021年4期)2021-04-20
汉字汉语研究(2019年2期)2019-08-27
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
中央民族大学学报(自然科学版)(2017年1期)2017-06-11
河北书画研究(2016年3期)2016-04-28
中国酿造(2016年12期)2016-03-01
中国酿造(2016年12期)2016-03-01
大连工业大学学报(2015年4期)2015-12-11
西南医科大学学报(2015年1期)2015-08-22
