
基于磁性CoFe2O4-石墨烯分子印迹技术联用高效液相色谱测定蜂蜜中的咖啡因
2018-11-07 06:24杭学宇张敏会
分析科学学报 2018年5期
王 露, 杭学宇, 宋 鑫, 王 芹, 张敏会
(淮安市疾病预防控制中心,江苏淮安 223001)
咖啡因为一种嘌呤生物碱,是日常饮料中的主要成分。咖啡因是中枢神经系统的兴奋药,对心血管系统具有正性作用,还能促进胃酸分泌等疾病。但若长期或大剂量摄入含咖啡因的饮料可能会引起高血压、休克等心血管系统疾病和中毒[1 - 2]。因此,有必要建立一种快速、准确、选择性高的检测食品中咖啡因含量的方法。
磁性分离作为一种在化学和医学领域[3 - 5]中发展起来的分离技术,在磁场的作用力下,根据物质的磁性差异,把磁性组分从非磁性组分中分离出来。磁性纳米粒子通过表面改性或表面共聚,能与无机材料或高分子聚合物或有机物相结合形成核壳结构复合微球,既具有磁性,表面又具有活性基团,能够进一步修饰。将分子印迹聚合物作为固相萃取材料,具有成本低廉、制备容易、耐热、耐强酸强碱等优点,已经广泛应用[6 - 9]。其中表面印迹使印迹的结合位点尽可能在印迹聚合物的表面,不但会使模板分子更容易接近结合位点有利于模板分子的洗脱,而且会增加分子印迹聚合物的印迹识别位点[10]。同时,在外加磁场的作用下,磁性材料能够很简便地与溶液分离,具有分离效率高和操作简便的优点。
本文成功合成以CoFe2O4-石墨烯为载体,多巴胺为功能单体和交联剂,咖啡因为模板分子制得的磁性分子印迹材料。建立磁性固相萃取结合高效液相色谱-紫外检测法(HPLC-UV)定量分析蜂蜜中的咖啡因。结果表明,建立的方法不但省时、高效、环保,而且方法的回收率、富集倍数、检出限等均令人满意,能够满足快速测定样品中咖啡因的要求。
1 实验部分
1.1 仪器与试剂
TECNAI-12透射电子显微镜(荷兰,The Netherlands);EV7型振动样品磁强计(美国,ADE公司);KQ218型超声波清洗器(昆山市超声仪器有限公司);pHS-25型酸度计(上海雷磁仪器厂);圆形磁铁(20×5 mm)。Aglient高效液相色谱仪(美国,Agilent公司):TC-C18色谱柱(150×4.6 mm,5 mm),流动相为甲醇-水(40∶60,V/V),流速0.5 mL/min,检测波长274 nm,进样量5 μL。
咖啡因、石墨粉末、甲醇、乙醇、乙酸、Fe(NO)3·9H2O、Co(NO3)2·6H2O、H2O2、KMnO4、H2SO4、H3PO4,均为分析纯,购于国药集团。0.1 mol/L的Tris-HCl缓冲溶液(pH=8.5);多巴胺溶液(10 mmol/L):称取0.1896 g多巴胺,溶于水中并定容至100 mL。实验用水为二次去离子水。
1.2 CoFe2O4-GO的制备
1.2.1氧化石墨烯(GO)的制备按照文献方法[11]制备GO。制备步骤如下:称取3.0 g石墨粉,加入400 mL的混合酸(H2SO4+H3PO4,V/V=9∶1)中。搅拌10 min,缓慢加入18 g KMnO4后,加热至50 ℃并搅拌12 h。待反应结束后,冷却至室温,然后置于冰浴中再搅拌0.5 h。逐滴加入30%H2O2直至溶液变为亮黄色,静置过夜,弃去上层清液。依次用5%HCl和蒸馏水洗净后干燥,然后溶于水中超声1 h后高速离心,取下面沉淀烘干,即制得GO。
1.2.2CoFe2O4-GO的制备首先称取上述制备的1.0 g GO溶于250 mL水中,超声2 h。再称取1.72 g Fe(NO)3·9H2O和0.62 g Co(NO3)2·6H2O溶于25 mL水中,混匀后,逐滴加入超声过的GO悬浮液并剧烈搅拌,然后将混合液pH调至12.0。将混合液升温至80 ℃,加入10 mL水合肼继续搅拌直至溶液呈黑色,待溶液冷却至室温,依次用水和乙醇洗净,用磁铁分离,真空烘干,制备得CoFe2O4-GO。
1.2.3磁性分子印迹材料(CoFe2O4-GO@MIPs)的制备称取20 mg上述制备CoFe2O4-GO,然后加入20 mL 10 mmol/L多巴胺溶液,20 mL 0.1 mol/L Tris-HCl缓冲溶液(pH=8.5)和8 mg目标分子咖啡因,反应8 h。其中多巴胺既作为功能单体也作为交联剂自聚合在CoFe2O4-GO表面,制备印迹聚合物。用磁铁分离出固体产物,用水洗涤数次,真空干燥。模板分子用甲醇-乙酸(9∶1,V/V)混合液洗脱,直至检测不出模板分子为止。真空干燥后即得到咖啡因磁性分子印迹聚合物(MMIPs)。同时,非印迹聚合物(NIPs)在制备过程中除不加入目标物质咖啡因,其余步骤与印迹聚合物一致。
1.3 样品的吸附与富集过程
整个磁性固相萃取过程如下:于小烧杯中放入10 mg CoFe2O4-GO@MIPs,加入100 mL咖啡因标准溶液。超声10 min,然后用磁铁分离固液两相,倒出上层清液,加入2 mL水洗涤磁性分子印迹材料,用1.0 mL甲醇-乙酸(9∶1,V/V)混合液超声洗脱后,待HPLC定量测定。
2 结果与讨论
2.1 CoFe2O4-GO@MIP的透射电镜图
图1A和1B分别是CoFe2O4-GO和CoFe2O4-GO@MIPs的透射电镜(TEM)图像。由图中可以看出所有的粒子为纳米级,大小均匀,分散性均比较好。

图1 CoFe2O4-GO(A)和CoFe2O4-GO@MIPs(B)的透射电镜(TEM)图Fig.1 TEM of CoFe2O4-GO(A) and CoFe2O4-GO@MIPs(B)
2.2 磁性分析

图2 CoFe2O4(a)、CoFe2O4-GO(b)和CoFe2O4-GO@MIPs(c)的磁滞回线Fig.2 VSM magnetization curves of CoFe2O4(a),CoFe2O4-GO(b) and CoFe2O4-GO@MIPs(c)
图2a、2b和2c分别是CoFe2O4、CoFe2O4-GO和CoFe2O4-GO@MIPs的磁滞回线曲线,三条磁滞曲线的形状相似,CoFe2O4的饱和磁化强度为29.1 emu/g,而包裹了石墨烯层后,饱和磁化强度下降为24.6 emu/g,再包裹分子印迹聚合物后,饱和磁化强度下降为22.0 emu/g,表明该磁性吸附剂在外加磁场的作用下具有一定磁响应特性。
2.3 实验条件优化
2.3.1功能单体本实验在甲基丙烯酸、丙烯酰胺、对乙烯苯甲酸、和多巴胺4种物质中选择功能单体。以咖啡因为模板分子,分别在CoFe2O4-GO表面以甲基丙烯酸、丙烯酰胺、对乙烯苯甲酸、和多巴胺4种物质为功能单体,分别合成4种磁性分子印迹材料。其形貌都呈有凹陷的纳米粒子,分别来做回收实验。实验结果表明甲基丙烯酸和多巴胺回收率达到90%以上,丙烯酰胺和对乙烯苯甲酸的回收率仅在75%左右。其中多巴胺作为单体也能作为交联剂,大大降低了有机试剂的污染。并且利用其可以自聚合在CoFe2O4-GO表面制备印迹聚合物,反应条件简单、易于控制印迹聚合物的形貌、结构。所以最终选择多巴胺为功能单体。同时实验考察了多巴胺和CoFe2O4-GO之间的比例对萃取效果的影响。实验结果显示,当两者质量比为2∶1时,聚合物材料对样品的吸附效果最好。
2.3.2模板分子浓度选择合成时模板的浓度对材料吸附性能有着重要影响。为了选择最佳模板分子浓度,实验考察了咖啡因在2~15 mg范围内的影响。随着聚合液中咖啡因浓度的增大,回收率增大。因为模板浓度越高,形成的印迹膜的识别位点就越多。当咖啡因浓度大于8 mg时,回收率减小,则有可能导致非特异性吸附增强,选择性降低。所以选择模板最佳质量为8 mg。
2.3.3吸附时间为了提高本方法的准确度和灵敏度,必须保证在吸附时间内使待测物最大程度进入到分子印迹的空穴中去。实验考察了时间为2~20 min对吸附的影响,随着时间的增加,回收率增大,当时间为10 min时达最大值之后保持平衡,故最佳时间选为10 min。
2.3.4CoFe2O4-GO@MIPs的用量为选择最佳CoFe2O4-GO@MIP用量,实验考察了用量2~15 mg对吸附效率的影响。结果显示,随着用量的增大,咖啡因的回收率迅速增大,用量10 mg时回收率最大。大于10 mg后回收率几乎保持不变,说明10 mg CoFe2O4-GO@MIPs足以吸附咖啡因。因此本实验选择吸附剂CoFe2O4-GO@MIPs用量10 mg。
2.3.5洗脱剂及其体积分别以甲醇-水(1∶2,V/V)、甲醇-水(2∶1,V/V)、甲醇-乙酸(9∶1,V/V)混合溶液作为本实验的洗脱剂。结果显示甲醇-乙酸(9∶1,V/V)混合液洗脱能力最好,这是由于乙酸破坏了咖啡因和印迹材料之间的氢键。为了进一步优化实验条件,对洗脱剂的体积也进行了考察。当洗脱剂体积大于1 mL,回收率不再增大。最后洗脱剂选择1 mL。
2.4 最大样品体积和富集因子
在10 mg CoFe2O4-GO@MIPs条件下,保持咖啡因的总量为5 μg,改变样品体积分别为10、20、50、100、150、200 mL进行吸附实验。实验结果如图3,当待测样品体积大于100 mL时咖啡因的富集回收率低于90%,因此富集咖啡因的最大试样体积为100 mL。由于1 mL甲醇-乙酸(9∶1,V/V)混合液能定量洗脱目标分子咖啡因,因此最大富集因子为 100。
2.5 CoFe2O4-GO@MIP吸附剂的稳定性
良好的磁性分子印迹材料不仅要有较高的吸附能力和特异的识别性,还要具有良好的稳定性。图4显示本实验中制备的CoFe2O4-GO分子印迹材料吸附剂能够重复使用超过100次。
2.6 吸附容量
称取10.0 mg制备的磁性分子印迹材料,加入至10 mL不同浓度(100、200、300、400、500 mg/L)的咖啡因标准溶液中,在室温下超声萃取5 h后离心分离,取上清液过0.45 μm滤膜,用HPLC法检测溶液中咖啡因浓度。同时做NIPs的吸附实验。根据吸附前后溶液中浓度的变化分别计算MIPs和NIPs对咖啡因的吸附容量。由上述静态吸附实验得磁性分子印迹材料的平衡吸附容量Qe与平衡浓度c的曲线如图5。随着咖啡因浓度的增大,吸附容量也逐渐增加,最后几乎保持不变,吸附达到平衡。MIPs和NIPs的饱和吸附容量分别为39、15 mg/g。MIPs的吸附容量是NIPs的2.6倍,说明MIPs识别空穴对模板分子具有特异性识别能力,因此具有较高的吸附能力。
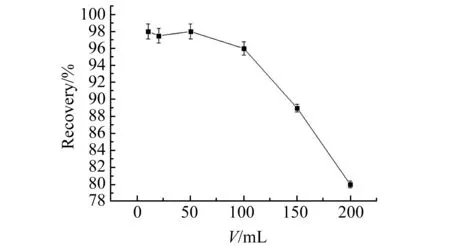
图3 样品体积对CoFe2O4-GO@MIPs吸附咖啡因的影响Fig.3 Effect of sample volume on the sorption of caffeine on CoFe2O4-GO@MIPs
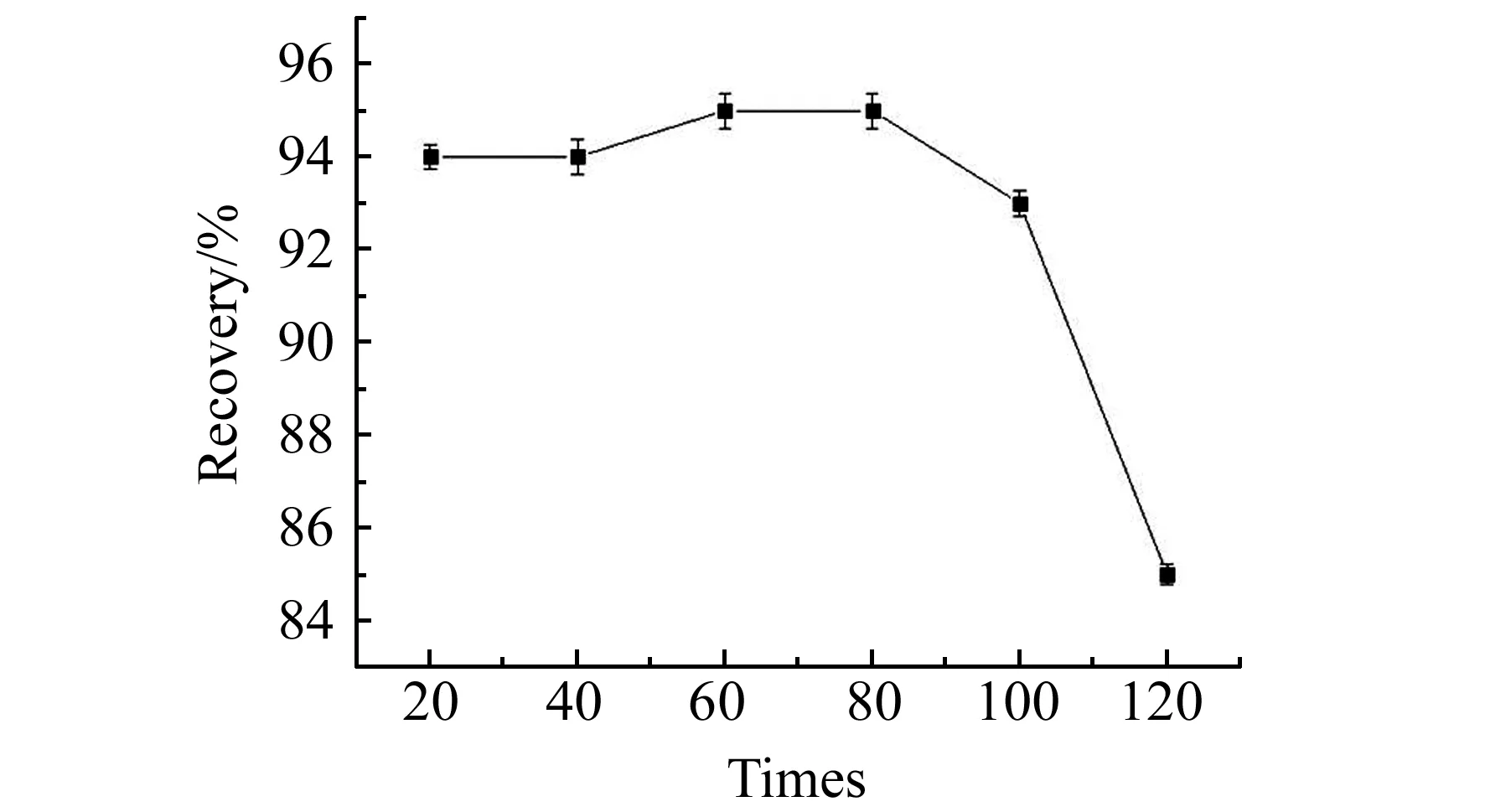
图4 吸附剂的重复性Fig.4 The repeatability of sorbent
2.7 CoFe2O4-GO@MIPs的选择性
在最佳实验条件下考察了CoFe2O4-GO@MIP在测定咖啡因时的抗干扰能力,结果如图6。选择了和咖啡因结构性质相似的物质:鸟嘌呤、次黄嘌呤、维生素B4、胞嘧啶、尿嘧啶、柠檬酸、诺氟沙星、环丙沙星、氧氟沙星、洛美沙星来进行选择性分析。经过咖啡因的CoFe2O4-GO@MIP处理的样品,咖啡因的吸附效率远远高于其他物质,而经非磁性分子印迹处理过的样品,其他物质吸附效率相差较小。可以看出本方法具有较高的识别效果。

图5 MIPs和NIPs对咖啡因的结合等温线Fig.5 Binding isotherms of caffeine onto the MIPs and NIPs

图6 咖啡因MIPs与NIPs吸附效率Fig.6 The adroption efficiency of MIPs and NIPs
2.8 分析特性
为了验证本方法用于定量分析的可行性,在最佳实验条件下,以0.1、1.0、5.0、10.0、20.0、50 μg/L标准溶液绘制外标校正曲线。考察了方法的线性范围、检出限、精密度、富集因子等。咖啡因在0.1~50 μg/L 范围内呈较好的线性关系,线性方程为:A=0.0936+0.0329c,相关系数R2为0.9987,以3倍信噪比(S/N=3)所得检出限为0.02 μg/L。用本方法对1 μg/L咖啡因标准液进行5次平行测定,相对标准偏差(RSDs)为4.7%,富集因子为100。表明本方法能满足实验选定目标分析物的检测要求,适宜实际样品的快速检测。
本实验采集超市里的蜂蜜等作为实际样品,取2 g样品溶于热水中,超声5 min,静置样品后过0.45 μm滤膜,备用上机。在优化条件下进行5次平行测定,同时做1、10 μg/L水平加标回收实验进一步评估方法的适用性和准确性。检测结果如表1所示,能满足痕量分析的要求。
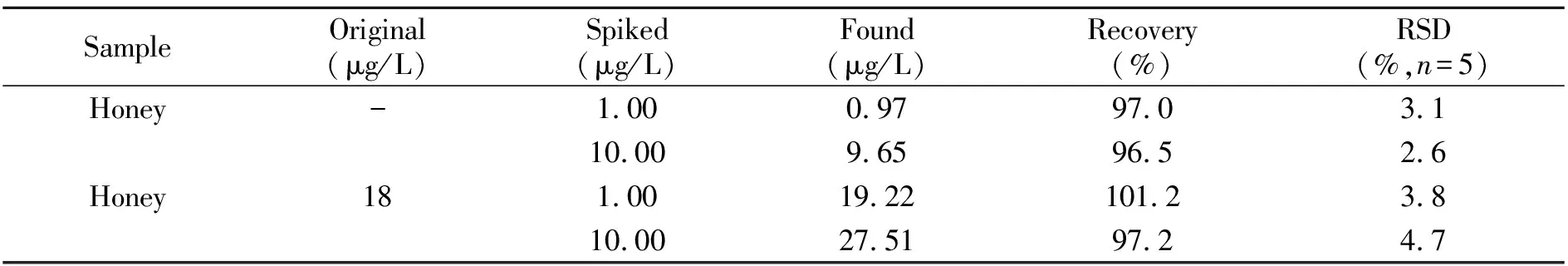
表1 蜂蜜样品中咖啡因的测定 (n=3)
3 结论
本实验采用CoFe2O4-石墨烯分子印迹技术与高效液相色谱联用技术,建立了一种快速测定实际样品中咖啡因新方法。实验结果表明,CoFe2O4-石墨烯分子印迹联用高效液相色谱法用于基质复杂的样品的测定可取得满意的结果,对于拓宽磁性分子印迹技术的应用范围具有重要意义。
猜你喜欢
陶瓷研究(2022年3期)2022-08-19
云南画报(2021年10期)2021-11-24
杭州化工(2020年2期)2020-08-31
小学生优秀作文(高年级)(2018年4期)2018-09-11
大自然探索(2017年10期)2017-10-28
军事文摘·科学少年(2017年4期)2017-06-20
大自然探索(2017年5期)2017-05-26
材料科学与工程学报(2016年2期)2017-01-15
中国摄影(2014年12期)2015-01-27
应用化工(2014年8期)2014-08-08
