
Influence of Photoisomerization on Binding Energy and Conformation of Azobenzene-Containing Host-Guest Complex
2018-10-19 08:00:56LIUPingyingLIUChunyanLIUQianMAJing
物理化学学报 2018年10期
LIU Pingying , LIU Chunyan , LIU Qian , MA Jing ,*
1 School of Materials Science and Engineering, Jingdezhen Ceramic Institute, Jingdezhen 333403, Jiangxi Province, P. R. China.2 School of Chemistry and Chemical Engineering, Key Laboratory of Mesoscopic Chemistry of MOE, Nanjing University, Nanjing 210023, P. R. China.
Abstract: The construction of a photo-controllable artificial molecular machine capable of realizing the light-driven motion on a molecular scale and of performing a specific function is a fascinating topic in supramolecular chemistry. The bistable switchable molecule, azobenzene (AZO), has been introduced into the supramolecular architecture as a key building block, owing to its efficient and reversible trans (E)-cis (Z)photoisomerization. The binding strength of the dibenzo[24]crown-8 (DB24C8) host and dialkylammonium-based rod-like guest consisting of an AZO moiety and the Z→E photoisomerization process in an interlocked host-guest complex have been investigated by the density functional theory (DFT) calculations and the reactive molecular dynamics (RMD)simulations by considering both torsion and inversion paths. The strong host-guest binding strength provides a necessary premise to stabilize the complex during the E-Z photoisomerization of the AZO unit, which is a terminal stopper to control the directional motion of the guest. A stronger binding strength for the Z isomer can be induced by the stronger hydrogen-bonding interaction. The steric effect is introduced into the Z isomer to force the ring slipping exclusively over the cyclopentyl terminal (pseudostopper). The host-guest complexation has a slight effect on the conformation of the AZO functional subunit for the two isomers. The faster Z→E photoisomerization process within the picosecond timescale is kinetically more favored than the dethreading of the ring through the pseudostopper subunit of the rod. After isomerization,a structure relaxation is observed for the crown ether ring within 500 ps. The flexible backbone of the crown ether ring is helpful in realizing steady and stable host-guest recognition during photoisomerization. Moreover, the orthogonality of the site-specific binding interaction is revealed by the similar binding energies obtained at similar hydrogen bonding recognition sites for various interlocked host-guest supramolecular systems although the constituents of the guests are different from each other. The introduction of two stereoisomers of the AZO subunit has little influence on the other conformations of guest subunits. These results are useful for the rational design of more sophisticated stimuli-controlled artificial molecular machines.
Key Words: Photoisomerization; Reactive molecular dynamics model; Azobenzene; Nanomotors;Pseudorotaxane; Supramolecular chemistry
1 Introduction
A host-guest system refers to a chemical system that is composed of two or more molecular components self-assembled together to form a supramolecular complex.Nowadays, the host-guest functional system is moving forward to the constitutional dynamically movable system in a controlled manner1,2with the potential application in fabrications of artificial interlocked nanostructures, such as molecular rotors, shuttles, muscles, pumps, and autonomous motors, etc.3–11. The challenge is the rational design of the movable building units to initiate and perform the movement of various nanomachines. A feasible strategy is to incorporate into supramolecular system a functional subunit that can alter its conformation when activated by external stimuli to realize the control of motion on the molecular scale12–16. Among the various external stimuli, light irradiation is frequently employed because it is cheap, convenient, and clean.Azobenzene (AZO) is one of the most widely used optical switching units owing to its high efficiency and excellent reversibility in a cis/trans isomerization upon the light excitation with appropriate wavelength17. The first use of azobenzene in supramolecular compounds was reported in 197918. Since then, the photoswitchable AZO-based supramolecular systems with the bistable trans (E) and cis (Z)conformations have attracted much attention for controllable modulation in numerous applications19. In this context, the design of light-driven host-guest complex represents an important step for the construction of smart molecular motors.Credi and co-workers designed a novel photoactive host-guest complex with an asymmetric rod terminated by an azobenzene and a cyclopentyl pseudostopper at each end through a crown ether, which could process repetitive directionally controlled molecular motion by alternating UV- and visible-light irradiation (Fig. 1)20,21. Thus, it is essential to clarify how photoisomerization of AZO functional subunits affect the mechanical motion and further to summarize the structure/property relationship to guide the design of more sophisticated artificial nanostructures.
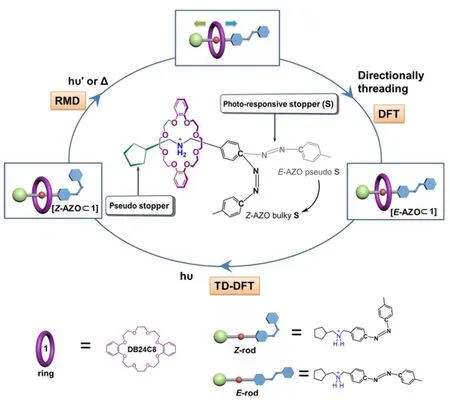
Fig. 1 Schematic representation of the photoactive host-guest system.
The theoretical investigation is a powerful tool to explore the fundamental understanding of switching behavior of azobenzene and its derivatives22–40. In our previous work, the light-induced E-Z isomerization of azobenzene-based nonlinear optical material (D-π-A type chromophore) was demonstrated to complete within femtosecond timescale in extremely dilute solutions22. We also simulated the collective photodriven E-Z switching process of azobenzene molecules on surface or in solutions using the reactive molecular dynamics (RMD)model23–29. Recently, Credi and co-workers reported a computational study on the ring-rod interactions as a function of the degree dethreading for a photoactive interlocked host-guest system with a symmetric rod terminated by two AZO end subunits by adopting a combination of first-principles molecular dynamics, metadynamics and quantum-chemical geometry optimization approaches30. In this work, we will simulate the Z→E photo-controlled switching behavior of a host-guest system with an asymmetric rod terminated by an AZO and a cyclopentyl pseudostopper at each end20,21(Fig. 1)by using RMD simulation with modified force fields, which considers both C―N=N―C torsion path and C―N=N inversion path of AZO isomerization. The influence of azobenzene subunit and its isomerization process on host-guest recognition and conformational change will hence be surveyed in such a supramolecular system.
On the other hand, we have recently built a fragment library consisting of 120 host-guest parings and it was found that the molecular recognition of interlocked host-guest architecture is largely dominated by size compatibility and electrostatic interaction between threadlike unit and macrocyclic unit41,42.When the different guest units with the same recognition subunit (binding site) are tightly bounded to the same host unit,their binding energies are nearly the same. Similar phenomenon has also been observed in a host-guest system with three different binding sites43, indicating the generality and orthogonality of site-specific binding interaction in our supramolecular library. It is interesting to check whether the new member of a photoresponsive host-guest functional system which has two stable stereoisomers: [Z-AZO⊂1] and[E-AZO⊂1] will have some new features to bring some new chances in rational construction of novel light-powered molecular machines.
2 Computational details
2.1 DFT calculations
The input configurations of the [Z-AZO⊂1] and [E-AZO⊂1]complexes and of their components ring 1, Z-rod and E-rod were constructed based on the reported crystal structure with the same recognition site (―NH2+―) and ring (DB24C8)44.The geometry optimizations were carried out by using the density functional theory (DFT) method at M06-2X/6-31G(d,p)level with the Gaussian 09 program45. The minimum structures of [Z-AZO⊂1] and [E-AZO⊂1] were selected for calculations of binding energies. The generalized energy-based fragmentation (GEBF) method46–48was used to calculate the MP2 (second-order Møller-Plesset perturbation theory) binding energy of large-size host-guest complex. It has been demonstrated that the binding energy calculated by M06-2X functional was quite close to the GEBF-MP2 result43. The binding energy, Ebwas calculated with M06-2X functional from the following expression,
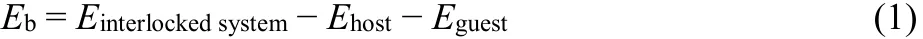
where Einterlockedsystemis the total energy of the interlocked compound, and the individual energies of ring and rod are denoted by Ehostand Eguest, respectively. A more negative value of Ebmeans a stronger binding interaction between host and guest. The basis set superposition error was computed by the counterpoise method49,50.
2.2 Reactive molecular dynamics (RMD) simulation
Since there is a relatively high energy barrier for the Z-E transition (> 134 kJ·mol−1) of AZO-containing host-guest complex, the conventional force fields based on MD simlations are difficult to describe the reverse Z–E conversion process.The photoisomerization between [Z-AZO⊂1] and [E-AZO⊂1]isomers can be completed through the cooperation between the out-of-plane torsion path of C―N=N―C dihedral angle and the in-plane inversion path of C―N=N bend angle. However,other existing reactive MD techniques, such as ReaxFF(reactive force field)51,52, REBO (a reactive empirical bond order)53and COMB (charge-optimized many-body)54,55potentials, which are mainly used in the bond cleavage/formation chemical reactions on basis of bond order variations. In order to simulate the dynamic cis-to-trans or called Z→E isomerization process of azobenzene terminal of thread, a RMD model23–26with the modified polymer consistent force field (PCFF) framework and a switching probability was performed. The RMD force field parameters considering the torsion path and inversion path of AZO moiety are modified according to the potential energy curves of E isomer and Z isomer calculated at QM level. The detail is illustrated in Section 3.2.1. The RMD simulations were implemented on the Discover module of Materials Studio package56. The canonical (NVT) ensemble was selected, and the temperature was controlled at 300 K by using a Nose-Hoover thermostat (with Q ratio of 1.0). The non-bonded van der Waals and Coulomb interaction were treated using the atom-based and Ewald truncation methods, respectively, and the cutoff distances for both nonbonded interactions were set to be 1.55 nm. The equations of the motion were integrated by the velocity Verlet algorithm with a time step of 1 fs. The production run of RMD simulation is 8 ns.
3 Results and discussion
3.1 Influence of azobenzene isomerization on host-guest recognition
The widely applied dibenzo[24]crown-8 host (ring 1) is selected in this work. The AZO-containing bistable host-guest systems with [Z-AZO⊂1] and [E-AZO⊂1] different conformations are compared with the previously studied host-guest compounds41,42, which are formed by a dialkylammonium-based rod-like guest with different neighboring groups and terminal stoppers. Fig. 2 illustrates the structure formulae of the threadlike guests (labeled as A, B, C,and D) and the interlocked host-guest systems (i.e., [A⊂1],[B⊂1], [C⊂1], and [D⊂1] ). The calculated host-guest binding strengths (−Eb) for the six systems bearing the same binding site, ―NH2+―, are shown in Fig. 2, from which the strong donor-acceptor parings between dialkylammonium and the crown ether were displayed with absolute values of binding energies larger than 209 kJ·mol−1. The orthogonality of site-specific binding interaction in host-guest architecture revealed in the fragment library is also applicable in the selected bistable [AZO⊂1] host-guest system. Specifically, the absolute value of Ebof [Z-AZO⊂1] is higher than that of[E-AZO⊂1] by about 42 kJ·mol−1. To further understand the impact of AZO terminal in threadlike subunit on the host-guest binding strength, we analyze the geometrical factor and the host-guest binding affinities for the two isomers, as shown in Fig. 3.
Little influence of ring-rod complexation on the AZO configuration is displayed in Fig. 3a, in terms of the C―N=N―C dihedral angles, φ, of azobenzene moiety. This also reflects the orthogonality in functional subunits for constructing host-guest systems.
A geometry requirement to construct a stable interlocked topology is that the cavity of ring (inner diameter of the macrocyclic host 1 is 0.71–0.82 nm) should be large enough to enclose the threadlike guest (the size of E-AZO pseudostopper:0.43 nm; cyclopentyl pseudostopper on the other end: 0.42 nm). The size of E-AZO unit is much smaller than the inner diameter of the cavity, thus there is no steric hinderance for rod movements. This is a crucial prerequisite to realize the directional controlled transit of a ring along an axle.
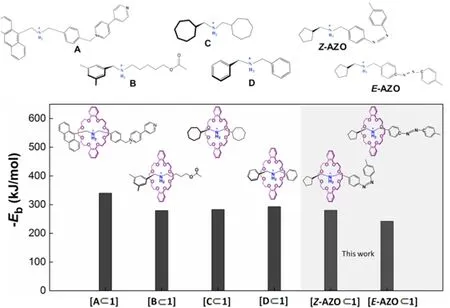
Fig. 2 Comparison of the calculated binding energies of six host-guest compounds with the similar binding sites.
Starting from the initial E isomer, the subsequent E→Z photoisomerization generates a nonsymmetric molecular axle in Z isomer. The size of Z-AZO bulky stopper is significantly increased to 0.94 nm, which is larger than the cage size of ring.Therefore, Z-AZO unit efficiently stops the shuttling of the DB24C8 ring. As a consequence, the photoinduced unidirectional threading and dethreading motion of a cyclic host along a threadlike guest would be obtained.
The electrostatic attraction and mutual affinity between host and guest has been addressed as one of the main driving forces to guide the supramolecular assembly41. The electrostatic potential (ESP) maps can visualize the 3D charge distributions of molecules. The electron-deficient (blue) and electron-rich(red) regions located in macrocyclic and threadlike components clearly illustrate their proton donor/acceptor functionalities involved in host-guest recognition. In general, the blue region is eager to be paired with the red region. The deeper color in ESP map means larger value of ESP, and the resultant interlocked compound is expected to have stronger binding strength. The ESP image-based approach was also applied to evaluate the site-specific hydrogen-bonding interaction dominated host-guest binding affinity42. In Fig. 3b, a strong hydrogen-bonding network (NH+…O) between the protonated amide group in rod and the polyether chain in ring is indicated to correlate with a high binding affinity of host-guest pair in[Z-AZO⊂1] than [E-AZO⊂1]. To shed further light on the difference of recognition property between the two isomers, the H-bonding interaction analysis was performed on optimized geometries for both Z and E isomers to estimate the binding strength. We estimated the number of hydrogen bonds (HBs)with the cutoff of H…O distance being set as 0.22 nm. As shown in Fig. 3b, the Z-AZO rodlike isomer in [Z-AZO⊂1]engages triple H-bonding interactions with DB24C8 ring(relevant H···O distance for two NH+…O HBs: 0.19 nm, 0.21 nm; CH…O HB: 0.22 nm). [E-AZO⊂1] only has two NH+…O HBs, in which the H…O distances (0.22 nm) of two HBs are longer than those of the two type HBs in [Z-AZO⊂1]. In molecular ESP maps, the deeper red color in cavity of ring for Z isomer means larger negative potential, i.e., stronger proton accepting ability. The calculated natural bond orbital (NBO)charges (in Fig. 3b) involved in the H-bonding interaction rationalize the above observation. Taken all together, it can be deduced that Z-rod should be a better binding affinity than E-rod with ring 1 to form interlocked architecture, suggesting that the stronger host-guest interaction in Z isomer than in E isomer. The results are consistent with the calculated binding energies for the two isomers. A higher affinity of host-guest recognition for Z isomer is required to achieve the thermodynamically stable interlocked skeleton while E→Z photoisomerization proceeds.
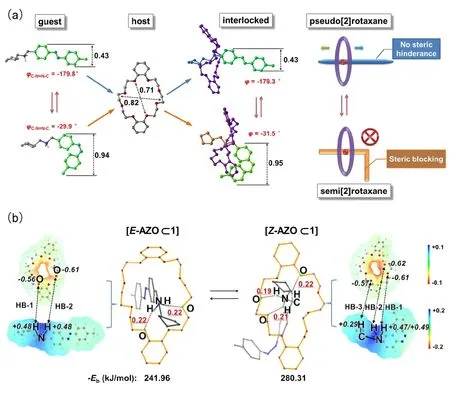
Fig. 3 (a) Optimized structures and geometrical parameters before/after host-guest interlocked process; (b) electrostatic potential maps (ESP) of the isolated rings and rods taken from the two isomers,and hydrogen bonding (HB) interactions for two isomers.The calculated natural bond orbital (NBO) charges involved in the HB interactions are also given.
3.2 RMD simulation of Z→E isomerization
3.2.1 Potential energy curves and force field functions of RMD model
To provide insight into the dynamic isomerization process of the interlocked system in the gas phase, RMD simulations considering both torsion and inversion path were carried out.For the RMD model, the key step is to determine the parameters of the force field by fitting the potential energy curves of the torsion path and inversion path of AZO isomerization. The details of the RMD method can be found in recent publications23–26. In the present work, the torsion and inversion energy curves of E-rod and Z-rod were calculated at(U)CAM-B3LYP/6-31G(d) level as shown in Fig. 4, which can reproduce the experimental UV-Vis adsorption spectrum26.According to the isomerization mechanism, the Z→E potential energy curve is assembled through two curves, that is, the first excited state (S1) curve from initial Z state to the transition state(TS) and the curve along the ground state (S0) from the transition state to the final E state, and verse vice. The modified function for Z→E potential energy curve based on polymer consistent force field (PCFF) in torsion pathway is written as

and E→Z potential energy curve is
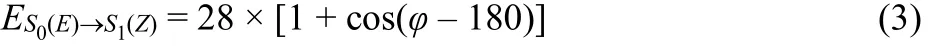
The fitted force field in inversion pathway for the ground state S0is expressed as

and the fitted potential curves for the first excited state S1of the two isomers are
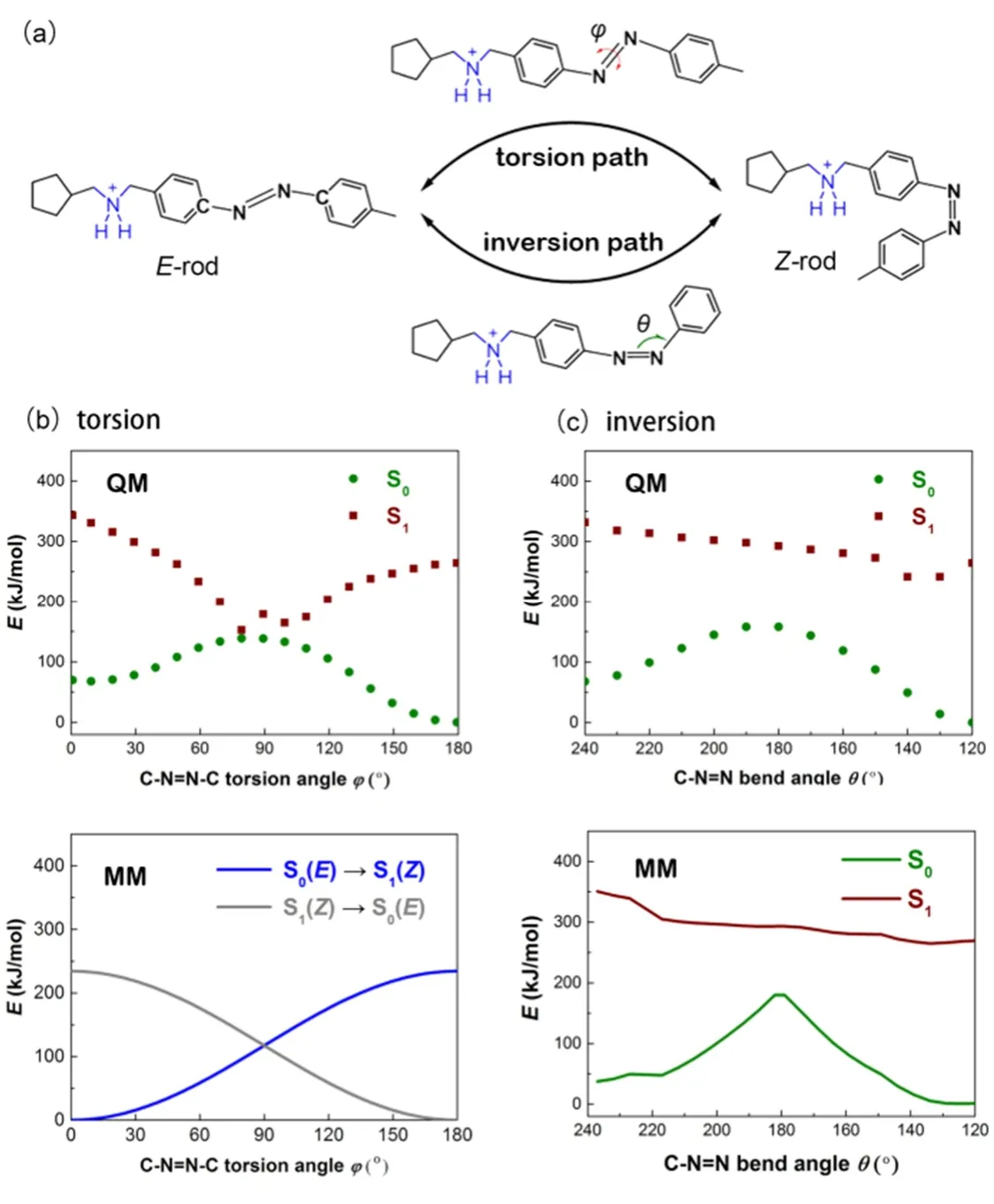
Fig. 4 (a) Schematic representation of the two different isomerization paths for host-guest system (the host was not displayed for clarity).The potential energy curves for Z/E isomerization of thread through (b) torsion and (c) inversion paths, respectively.
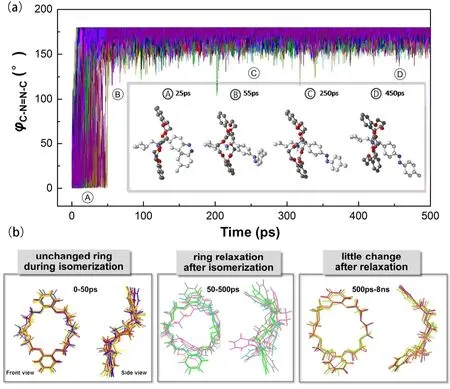
Fig. 5 (a) Fluctuations of the C―N=N―C torsion angle, φ,with the time evolution and the representative snapshots extracted from the RMD simulations; (b) overlay of ring conformations during and after AZO isomerization.
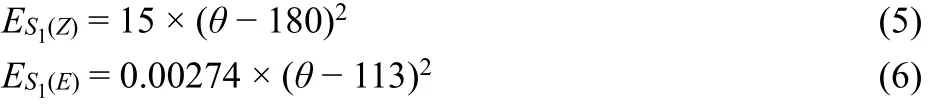
respectively. Based on the modified force fields, the Z→E isomerization process can be simulated for photoactive host-guest system.
3.2.2 Conformational dynamic changes during the Z→E photoisomerization
One hundred parallel 500 ps RMD simulations of Z→E switching process were performed in gas phase. The 100 RMD trajectories of dihedral angle of C―N=N―C (φ) within 500 ps time duration were displayed in Fig. 5a. Most AZO moieties in rods switch from the Z→E configuration within 50 ps. The representative four snapshots extracted from the RMD simulations clearly exhibit the dynamic switching process of AZO terminal (inset in Fig. 5a). Notably, no significant relative motion of the rod to the cavity center of ring is found in such a short time scale, holding the tight bound between dialkylammonium moiety in rod and crown ether ring. In addition, the other subunits, except for the photoswitchable AZO unit in axle, exhibit negligible difference during Z→E isomerization.
However, up to 500 ps RMD simulation, an apparent change in DB24C8 ring conformation is observed. To better understand the influence of switching behavior on the macrocyclic backbone, Fig. 5b illustrates the superimposed conformations extracted at every 10 ps from the early 0–50 ps(during isomerization) and at every 100 ps from the later 50–500 ps (after isomerization) RMD simulation, respectively.Different from the slightly fluctuation during isomerization(0–50 ps), the crown ether ring undergoes large geometric change and reorganization in 50–500 ps, indicating the flexibility of DB24C8 backbone. In other words, after the isomerization within 50 ps, the flexible crown ether ring will subject to conformational relaxation, in order to better host the final E-rod. The flexible backbone of ring provides the optimal orientation for the most favorable host-guest binding interaction, which should be strong enough to avoid the dethreading of E-rod. An 8 ns RMD simulation has also been carried out. The result shows that the conformation of ring has little change after structure relaxation (Fig. 5b). In addition,although the temperature of our RMD simulation is set at 300 K, the system temperature undergoes an obvious fluctuation with time evolution as shown in Fig. S1 of Supporting Information; for instance, the system temperature rose to 350 K at 50 ps.
It should be pointed out that the solvent may also bring some effects on the host-guest interactions. We have investigated the influence of polar solvent (CH3CN) on conformational changes and NMR signals associated with the host-guest recognition of pseudorotaxanes and [2]rotaxanes43,57. For the strongly bounded sites, such as the protonated amino ―NH2+― unit paired with the polyether chain, there is little solvent effect on1H NMR chemical shifts of the protons involved in host-guest recognition43. But for the weaker binding sites, such as the urea group paired with the same polyether chain, the dipole-dipole interactions between the polar CH3CN solvents and the polar bonds (C―H bond) in the host caused the directional and inhomogeneous distribution of solvents close to the flexible subunit of host57. The solvent effect on host-guest binding behavior during isomerization reaction will be discussed in the future.
4 Conclusions
In this work, the influence of bistable azobenzene subunit introduced to a host-guest functional system has been investigated by DFT calculations and RMD simulations. For the two isomers, the host-guest complexation slightly affects the conformation of azobenzene subunit, indicating the orthogonality of functional subunits. There is no steric hinderance for [E-AZO⊂1], thus the crown ring would pass over the E-AZO terminal to the ammonium recognition site to form a pseudorotaxane. Steric effect is introduced into the[Z-AZO⊂1] system to force the ring slipping exclusively over the cyclopentyl terminal. As a consequence, the directional movement of the threadlike unit through the macrocyclic unit has been accomplished. The two isomers have strong ring-rod binding strength. Due to the stronger H-bonding interaction,[Z-AZO⊂1] has a stronger host-guest binding strength than[E-AZO⊂1]. Based on the RMD model considering both C―N=N―C torsion path and C―N=N inversion path, the Z→E switching behavior of AZO-containing supramolecular system was simulated. The RMD results show that the photoisomerization completes within 50 ps, much faster than threading/dethreading process. The flexible ring host undergoes obvious conformational fluctuations after the isomerization.Such a picture can be extended to fabricate new artificial supramolecular system with photochromic compounds acting as light-driven units.
Supporting Information: available free of charge via the internet at http://www.whxb.pku.edu.cn.

- 物理化学学报的其它文章
- On the Simulation of Complex Reactions Using Replica Exchange Molecular Dynamics (REMD)
- Free Energy Change of Micelle Formation for Sodium Dodecyl Sulfate from a Dispersed State in Solution to Complete Micelles along Its Aggregation Pathways Evaluated by Chemical Species Model Combined with Molecular Dynamics Calculations
- Reaction Mechanisms in the Thermal Decomposition of CL-20 Revealed by Reax FF Molecular Dynamics Simulations
- Deformation of Polymer-Grafted Janus Nanosheet: A Dissipative Particle Dynamic Simulations Study
- Selective Permeation of Gas Molecules through a Two-Dimensional Graphene Nanopore
- Microscopic Investigation of Ethylene Carbonate Interface:A Molecular Dynamics and Vibrational Spectroscopic Study