
1,2,4-三氮唑构筑锌配位聚合物的合成、结构及性能研究
2018-10-17 06:09:00苗卫妮何婷婷
陕西科技大学学报 2018年5期
刘 冰,苗卫妮,何婷婷,雷 行,张 甜
(陕西科技大学 化学与化工学院,陕西 西安 710021)
0 引言
配位聚合物是无机金属离子与有机配体通过配位键连接形成具有空间有序结构的一类多维配合物,相对传统无机材料,具有结构可调性、通用性及柔韧性等优点[1].近年来,配位聚合物的研究取得了巨大的进步,已广泛应用于催化[2]、离子交换[3]、药物传递[4]、气体吸附与分离[5]、化学传感器[6]、发光器件[7,8]、物质检测[9-11]等研究领域.配位聚合物的结构组建及构效关系研究是配位化学的研究热点之一[12,13].在该领域开展的大量研究初步揭示所存在构效关系,并已运用于配合物的结构设计和控制合成[14-18].其中的一个设计理念是选择多功能的桥连配体,并依据配位聚合物可能的应用范围,选择与之匹配的无机金属中心,构筑具有特定性能的配位聚合物[19].含N/O有机配体,因其种类繁多和构型多样,为常用配体.1,2,4-三氮唑是一类富π电子的芳香性N-杂环化合物,易于构建一些高对称性的结构[20-22],其配合物已运用于磁性材料、光驱动分子器件、信息存储及有机蓝光材料等领域[23-27].1,2,4-三氮唑的主要官能团为三氮唑环,其配位方式主要有三种:μ3-1κN:2κN:4κN、μ2-1κN:2κN、μ2-1κN:4κN.1H-1,2,4-三氮唑构型简单,来源丰富,具有生色基团三氮唑环,作为构筑模块易于和金属中心自组装形成具有新颖结构和优异光电性能的配位聚合物[28].
本文以1H-1,2,4-三氮唑为配体,与Zn(OAc)2(OAc-=乙酸根离子)不同比例混合,水热合成3D配位聚合物[Zn(tr)(OAc)](1).通过X-射线单晶衍射(SCXRD)确定化合物1的单晶结构,X-射线粉末衍射(PXRD)确定测试样品的物相纯度.通过热重分析(TGA)、荧光光谱分别表征其热稳定性及发光性能.
1 实验部分
1.1 原料与仪器
(1)主要试剂:实验所用药品未做进一步纯化.Htr购自梯希爱(上海)化成工业发展有限公司;Zn(OAc)2和DMF购自国药集团化学试剂有限公司;全氟聚醚真空油(FOMBLIN Y LVAC 25/6)购自西宝生物有限公司,均为分析纯;红外光谱所用KBr为光谱纯,购自国药集团化学试剂有限公司.
(2)主要仪器及测试条件:傅立叶红外光谱(Bruker Tensor 27,布鲁克)采用KBr压片法,测量范围4 000~400 cm-1;热重分析仪(SDT Q600 V8.3 Build 101,美国TA仪器):样品在N2氛下测试,N2流速为20 cm3·min-1,升温速率为10 ℃·min-1,测量温度为20 ℃~800 ℃;X-射线粉末衍射仪(Rigaku D/Max-3c,日本理学株式会社),辐射源为Cu Kα射线(λ1=1.540 598 Å,λ2=1.544 426 Å),测试条件为室温下连续模式,工作电压为40 kV,工作电流为30 mA,2θ扫描范围为3 °-60 °,步进为0.02 °,扫描速度为4 °min-1;固态荧光光谱(Hitachi F-4600,日本日立株式会社),室温下测试,以氙灯作激发光源,发射和激发狭缝均为2.5 nm,步进为2.0 nm,积分时间为0.1 s.
1.2 [Zn(tr)(OAc)](1)的合成
0.2 mmol,0.013 8 g Htr,0.6 mmol,0.131 7 g Zn(OAc)2·2H2O与5 mL水混合,磁力搅拌10 min,置于25 mL聚四氟乙烯水热罐中,140 ℃加热4天,以5 ℃/30 min的降温速率至室温,获得无色片状晶体,依次用水和乙醇洗涤,50 ℃干燥24小时,收集产物14.4 mg,产率为37.4%.化合物1的FT-IR光谱数据(KBr,cm-1):3 436.5(m),3 159.9(m),3 122.0(s),3 008.3(m),2 792.5(w),2 571.3(w),2 451.4(w),2 362.4(m),1 780.0(m),1 514.7(s),1 425.7(s),1 343.7(m),1 286.5(s),1 159.6(s),1 077.6(s),1 027.3(m),995.6(s),932.2(w),900.5(s),675.4(s),615.8(m),514.5(m).
1.3 X-射线单晶衍射
使用全氟聚醚真空油在显微镜下挑选适合X-射线单晶衍射用的晶体.将挑选的晶体置于Bruker X8 Kappa APEX II CCD面探衍射仪上,使用APEX2软件包收集X-射线单晶衍射照片.室温下利用石墨单色化的Mo kα射线(λ=0.710 73 Å),以ω扫描方式收集一定角度的衍射数据.利用SAINT+程序进行衍射数据的指标化和还原,使用SADABS软件包对衍射数据进行洛仑兹和极化影响的强度校正及经验吸收.化合物1的晶体结构通过使用SHELXTLTM晶体软件包由直接法解出,并基于F2函数对非氢原子经全矩阵最小二乘法进行精修.所有非氢原子的热参数进行各向异性修正.氢原子由理论加氢得到,只计算不修正.化合物1的晶体学数据详见表1所示,主要键长和键角列于表2中.
化合物1的晶体学数据(不包含结构因子)已经上传到剑桥晶体学数据库中,CCDC号为1583756,可通过网址免费获取:www.ccdc.cam.ac.uk/data_request/cif.

表1 化合物1的晶体学数据
R1=(Σ||Fo|-|Fc||/Σ|Fo|).wR2=[Σ(w(Fo2-Fc2)2)/Σ(w|Fo2|2)]1/2
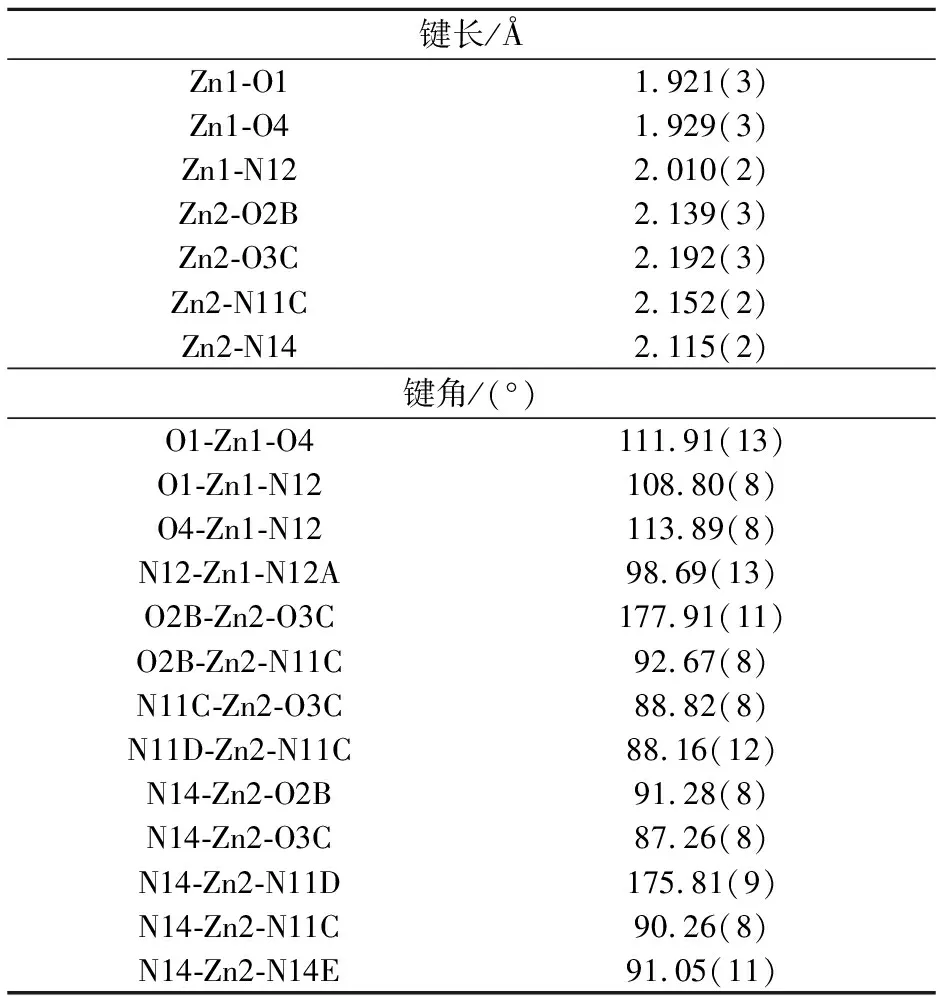
表2 化合物1的主要键长(Å)和键角(°)
对称码:A=-x,y,z;B=1/2-x,-2-y,1/2+z;C=1/2-x,-3-y,1/2+z;D=1/2+x,-3-y,1/2+z;E=1-x,y,z
1.4 拓扑分析
根据Wells,A F拓扑网络结点的定义[29]及化合物1的结构特征,[Zn(OAc)2]次级单元、Zn2和tr-配体分别虚拟为4-、6-、3-连接结点,通过拓扑结构分析软件包TOPOS 4.0中的ADS程序计算化合物1拓扑结构.
2 结果与讨论
2.1 合成
Zn(OAc)2与Htr的比例为1.0∶0.2,0.8∶0.2和0.6∶0.2时,在相似的反应条件下反应均得到化合物1,上述反应所得单晶,其单晶扫描所获晶胞参数(a=9.283 9,b=8.401 3,c=8.145 9,α=β=γ= 90 °)与化合物1的晶胞参数(a=9.272 9,b=8.372,c= 8.131 7,α=β=γ=90 °)一致.且各产物PXRD图谱与化合物1的PXRD图谱一致,表明在不同比例下,产物均为化合物1(如图1所示).

图1 化合物1的不同比例条件下的实验测试PXRD图样与单晶数据数据拟合PXRD图样比较
2.2 晶体结构
化合物[Zn(tr)(OAc)](1)是一个3D框架结构,由[Zn(OAc)2]次级单元连接相邻[Zn(tr)]2D层构建而成.其不对称单元包含有2个0.5占据的Zn离子,一个tr-配体及2个0.5占据的OAc-离子.2个0.5占据的Zn(II)离子上的2个正电荷被1个tr-配体和2个0.5占据的OAc-离子上的2个负电荷中和,化合物1保持电中性状态.四配位的Zn1与2个OAc-上的O1和O4,及2个tr-配体上N12和N12A(A= -x,y,z)连接形成一个畸变四面体(如图2所示).而Zn2的配位环境为六配位的八面体:其基面由4个tr-配体上N原子构成(N11C,N11D,N14,N14E;C=1/2-x,-3-y,1/2+z;D=1/2+x,-3-y,1/2+z;E=1-x,y,z);2个乙酸根氧原子O2B(B=1/2-x,-2-y,1/2+z)、O3C占据八面体的两个顶点(如图2所示).tr-配体和OAc-离子分别采用μ3-1κN:2κN:4κN和μ2-bidentate桥连方式(如图3所示).4个tr-配体通过1位和4位N原子交替连接4个Zn2离子,形成一个16元环.相邻16元环通过共边的方式连接形成一个沿ac平面格子状的[Zn(tr)]2D层,其拓扑结构为(4,4)2D层(如图4所示).2个OAc-离子以单齿模式连接Zn1形成[Zn(OAc)2]次级单元.该次级单元中的OAc-离子与Zn2连接,及Zn1通过与[Zn(tr)]二维层中tr-配体2位N原子连接,将相邻2D层连接形成3D框架结构(如图4所示).
根据化合物1的结构特征,通过TOPOS 4.0拓扑结构软件分析包,将[Zn(OAc)2]次级单元、Zn2离子和tr-配体分别虚拟为4-、6-、3-位节点.其点符号分别为{32.4.73},{32.4.76.86}和{3.72},因此化合物1的整体结构可简化为一个(3,4,6)连接的3,4,6T56拓扑网络,其点符号为{3.72}2{32.4.73}{32.4.76.86}(如图4所示).
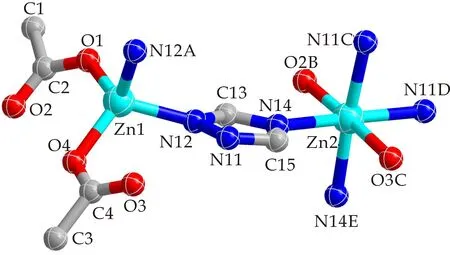
图2 化合物1中Zn(Ⅱ)原子的配位环境图(对称码:A=-x,y,z;B=1/2-x,-2-y,1/2+z;C=1/2-x,-3-y,1/2+z;D=1/2+x,-3-y,1/2+z;E=1-x,y,z)
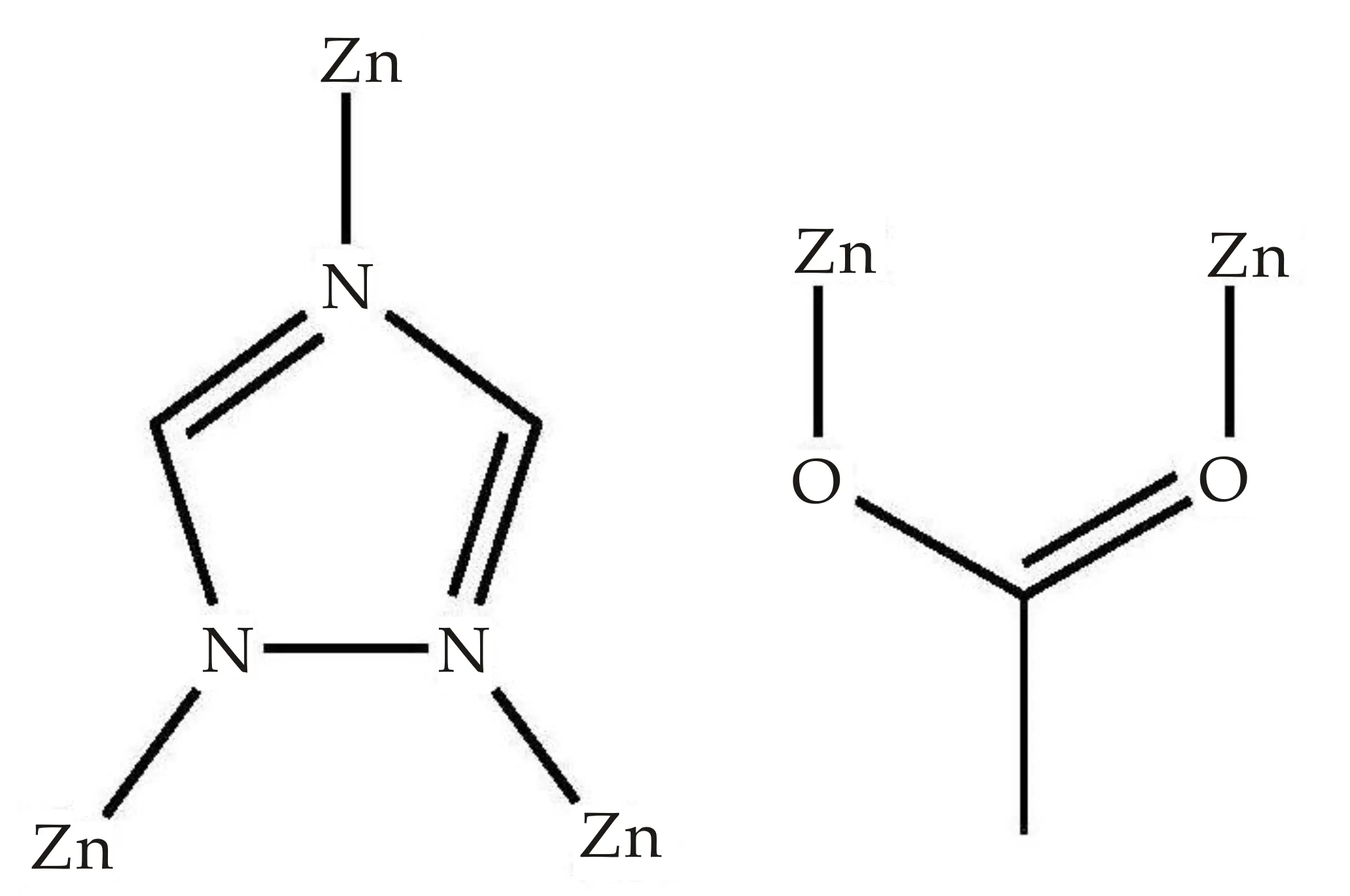
图3 tr-配体和OAc-离子的配位方式图
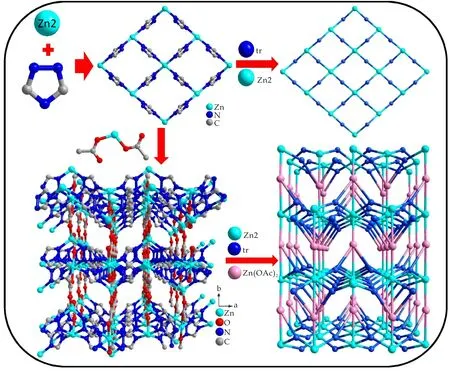
图4 化合物1的结构构建图及拓扑结构图
2.3 性能表征
2.3.1 X-射线粉末衍射
化合物1的PXRD图样与通过单晶数据拟合的PXRD图样一致,表明测试样品为化合物1的单一物相,性能测试样品与单晶样品一致,亦表明单晶结构模型解析与化合物1实际结构相符(如图1所示).
2.3.2 固态荧光光谱
Htr为富π电子N杂环,可发生配体内的π*→π跃迁,在有机蓝光材料方面有潜在应用.化合物1和Htr室温固态荧光光谱如图5所示.在270 nm紫外光激发下,化合物1在369 nm处有一宽的发射峰.Htr配体在360 nm紫外光激发下,产生427 nm浅蓝色荧光,归属于配体内的π*→π跃迁(ILCT).与Htr发射光谱比较,化合物1的荧光发生了约58 nm的蓝移.已报道的1,2,4-三氮唑配位聚合物的发射峰常见于420~460 nm,归属于ILCT或配体-金属电荷跃迁[30-34].与文中化合物1完全不同.化合物1在427 nm处的荧光发生淬灭,表明1,2,4-三氮唑环上π*激发电子没有回到基态,而是传递给OAc-,发生OAc-→Zn跃迁.
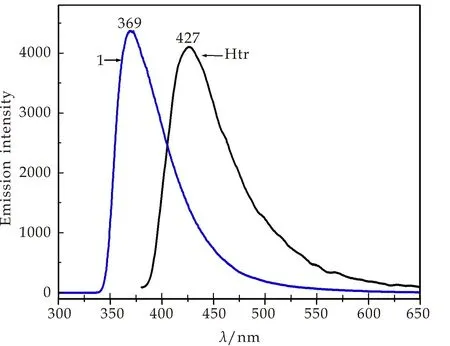
图5 化合物1和Htr配体的荧光光谱图
2.3.3 热重分析
化合物1的热重分析表明在211 ℃之前,化合物1保持稳定,未出现失重现象.其失重分为二步:第一步出现在211 ℃~476 ℃,失重较小,约3.54%;第二步失重发生在476 ℃之后,直到541 ℃失重约54.61%.211 ℃~541 ℃两步失重共计58.15%,归属于tr-配体和OAc-离子部分分解(OAc-分解为CH3CO-).实际失重58.15%与理论计算值57.68%非常接近.乙酸根残余O原子作为氧化剂与Zn(II)离子反应生成ZnO,残余物比重为41.85%,与以ZnO为残余物的计算值42.27%接近(如图6所示).化合物1的热稳定性相较其他1,2,4-三氮唑配合物的热分解温度(300 ℃左右),略有降低,与OAc-稳定性差有关.
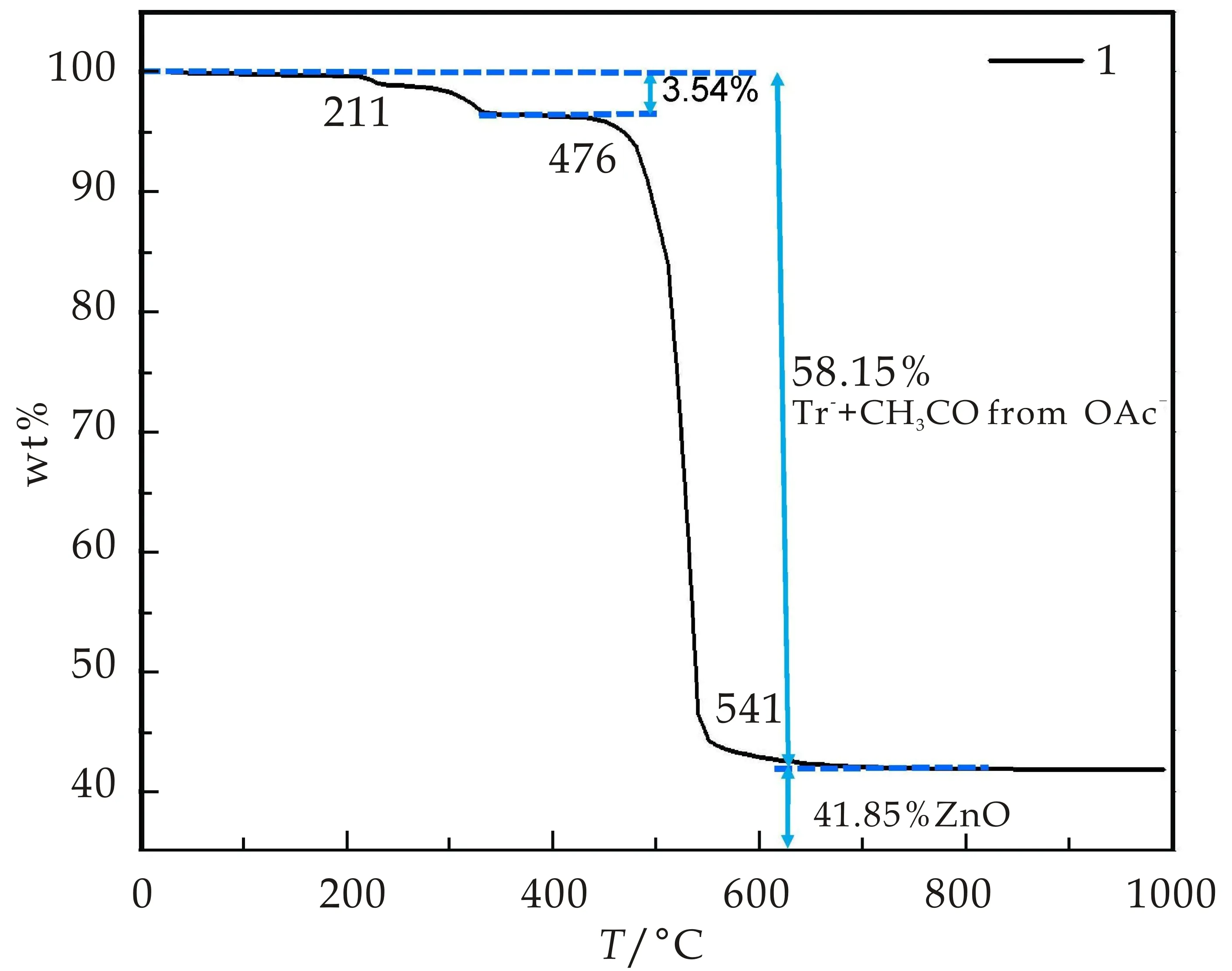
图6 化合物1的热重曲线图
3 结论
本文通过不同比例的Zn(OAc)2与Htr配体水热合成3D配位聚合物[Zn(tr)(OAc)](1).其3D框架结构由[Zn(tr)]2D层与[Zn(OAc)2]次级单元交织而成.该化合物的拓扑结构分析表明,将[Zn(OAc)2]次级单元、Zn2和tr-配体分别虚拟为4-、6-、3-位节点,化合物1简化为(3,4,6)连接的3,4,6T56拓扑网络,其点符号为{3.72}2{32.4.73}{32.4.76.86}.PXRD的测试图样证实:不同反应物比例获得的产物均为化合物1;PXRD测试图样与由单晶数据拟合的PXRD图样一致,表明样品性能测试的样品与单晶结构测试样品物相同.TG分析表明化合物1的失重分为两步:在211 ℃之前,化合物1保持稳定;第一步发生在211 ℃~476 ℃,第二步发生在476 ℃~541 ℃.两步总计失重58.15%,归属于tr-配体和OAc-分解为CH3CO-,与理论值57.68%非常接近.固态荧光光谱表明化合物1在270 nm光激发下,在369 nm产生一较宽的发射峰.与Htr固态荧光比较,有约58 nm蓝移,表明该发射峰与1,2,4-三氮唑环的π*→π跃迁无关,可能归属于OAc-→Zn跃迁.
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09 06:12:08
当代陕西(2019年6期)2019-04-17 05:04:10
中国科技信息(2016年19期)2016-10-25 08:15:06
中国科技信息(2016年6期)2016-08-31 07:27:16
项目管理技术(2016年6期)2016-05-17 05:39:02
中国科技信息(2015年24期)2015-11-07 08:52:23
中国科技信息(2015年23期)2015-11-07 08:25:56
技术与教育(2014年2期)2014-04-18 09:21:39
江南大学学报(人文社会科学版)(2014年3期)2014-02-28 17:45:44
无机化学学报(2014年4期)2014-02-28 17:31:08
