
端锚聚合酶作为药物靶点的研究进展
2018-09-08 03:05尹秀山张万忠
中国药理学通报 2018年9期
李 根,赵 栋,李 健,3,尹秀山,4,张万忠
(1.沈阳化工大学制药与生物工程学院,辽宁 沈阳 110142;2.西安医学院基础与转化医学研究所,陕西 西安 710021;3.肯塔基大学药学院,美国 列克星敦 40536;4.卡罗林斯卡医学院微生物,肿瘤与细胞研究所,瑞典 斯德哥尔摩 17165)
端锚聚合酶(tankyrase,TNKS),包括TNKS1和TNKS2,属于聚腺苷二磷酸核糖聚合酶[也称作ADPribosyltransferase,ARTDs或poly(ADPribose)polymerases,PARPs]家族,人源PARP家族包括17个成员[1]。该家族具有1个共有的催化结构域——PARP,其催化底物NAD+的ADP-核糖单位,将之加入到靶蛋白的谷氨酸或赖氨酸残基上。该家族成员PARP1研究的比较深入。PARP1参与多种细胞进程,包括DNA修复、复制和转录调控。PARP1可感应DNA损伤,并形成单链缺口(single-strandbreaks,SSB),可以作为DNA修复的桥梁发挥作用,因此与多种肿瘤密切相关[2]。针对PARP1的多个小分子抑制剂被研发并进入临床Ⅲ期试验,如维利帕尼、BMN-673,以及临床前期的候选药物如E7016、NMS-P118[3]。这些药物的初步成功证明了药物靶向TNKS的潜力。近年来,更多研究集中于各个PARP家族成员的详细结构特征,以及高度选择性小分子抑制剂的设计。TNKS作为有潜力的药物靶点,成为靶向研究的重点。本文总结了TNKS作为药物靶点的分子基础和TNKS抑制剂开发领域的最新进展,并展望了TNKS作为药物研发靶点的前景。
1 TNKS的结构
TNKS1(tankyrase1/ARTD5/PARP5a)和TNKS2(tankyr-ase2/ARTD6/PARP5b)与该家族其它成员相比,含有2个独特的结构域:介导蛋白质相互作用的锚蛋白重复序列簇(ANK, ARCs)和形成同源或异源多聚体的SAM结构域(Fig 1A)。两种酶蛋白序列高度保守,同源性高达80%。位于蛋白的C端的催化结构域在两者之间的序列同源性达89%。4个ARC都具有1段保守序列RXXPDG,该序列介导蛋白质相互作用[4]。由SAM区域介导的TNKS多聚化可被自体聚ADP-核糖基化而阻断,这使得TNKS成为一类可根据PARs水平而组装或者分解的一类蛋白质[5]。催化结构域包含2个结合位点:结合并水解NAD+的供体位点和容纳准备修饰聚ADP-核糖基化靶蛋白的受体位点。其中,供体位点分为结合在烟酰胺部分和腺苷酸部分的亚结合位点[6]。在TNKS1中,D环通过疏水作用关闭在烟酰胺位点附近的结合槽;而在TNKS2中,主要通过His1048关闭在腺苷酸位点附近的结合槽。当NAD+与位点结合或者有抑制剂存在时,D环会打开[7]。尽管D环在TNKS1和2的氨基酸序列是相同,但是两者构型并不相同(Fig 1D、1E)。
2 TNKS的生物学功能
TNKS分布在细胞的多个亚器官。在人类细胞中,TNKS在端粒、核孔、高尔基复合体、细胞质、细胞膜等都有发现。与其广泛分布相对应,TNKS在细胞水平参与多种调控,例如端粒的稳态平衡、有丝分裂、Wnt信号通路、葡萄糖吸收(Fig 2)[8-9]。特别是对Wnt信号通路的作用,多种疾病中发生Wnt信号通路的过度激活[10],Wnt通路参与炎症方面的机制逐渐被证实[11]。目前的模型预测,在存在功能性降解复合物的情况下,TNKS通过增加Axin的降解,降低了降解复合物的稳定性,这导致β-catenin降解减少,从而增加Wnt信号的水平。通过小分子抑制TNKS的催化活性,可稳定Axin,进而调控Wnt通路。靶向Wnt信号通路是晚期直结肠癌Wnt依赖性治疗APC突变的一个非常有吸引力的策略,因此,TNKS有希望成为调控直结肠癌中Wnt通路的治疗靶点。虽然TNKS1和TNKS2具有功能冗余性,但其亚细胞定位和蛋白质-蛋白质相互作用的差异可能导致两者具有独特的功能。该方面尚待进一步的研究。
3 TNKS抑制剂
像其他PARPs一样,TNKS抑制剂靶向于催化结构域。利用Wnt通路的荧光素酶报告基因筛选发现了第1种TNKS酶选择性抑制剂IWR-1和XAV939[12]。基于同样的筛选,也发现了TNKS特异性抑制剂WIKI4、JW55、JW74及其衍生物G007-LK等[13]。生物化学方法与基于片段的配体设计的结合最近被用于筛选和表征化合物来抑制TNKS,例如新型抑制剂E7449能够同时抑制PARP1/2和TNKS1/2,在BRCA缺陷的体内模型中显示出有效的抗肿瘤活性,并增加临床前化疗的效果。E7449对TNKS1/2的抑制与传统的抑制剂有明显区别,其可以扩大缺乏DNA修复能力的肿瘤以外的潜在治疗应用。通过对Wnt通路抑制剂的深入研究,可能会发现一些Wnt通路和TNKS的双重抑制剂。Gustafson等[14]最新的研究结果表明,Wnt通路抑制剂FH535使Axin2的PARs修饰减少,表明其同样可以阻断TNKS1/2酶活性,其可以作为控制骨肉瘤的有效化疗抑制剂。随着TNKS作为药物靶点对许多疾病(包括选定的肿瘤细胞)所展示出的良好效果,针对TNKS新型高效的抑制剂相继被研发出来,在以下部分中,我们将根据TNKS抑制剂的作用位点及作用方式,对各种抑制剂进行总结。
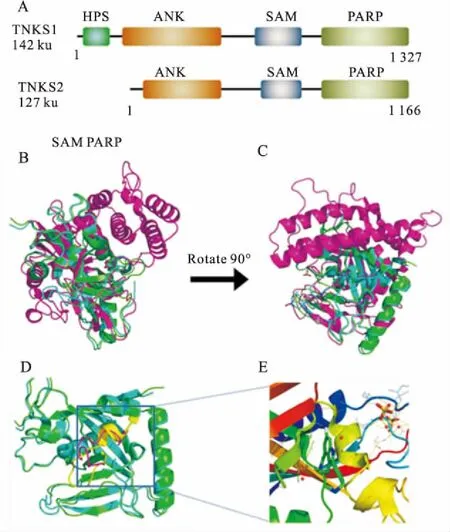
Fig 1 Structure of TNKS
A: The domain schematic of anthropogenic protein TNKS; B, C: The crystal structure overlay of TNKS1, TNKS2 and PARP1(TNKS1 identified as green, TNKS2 identified as cyan, PARP1 identified as purple, and C is the conformation of B rotated 90°; D,E: The crystal structure of D-loop(TNKS1 identified as purple, TNKS2 identified as yellow, E is the enlarged crystal structure of TNKS2 D-loop).
A: TNKS interacts with TRF1 to maintain telomere homeostasis; B: TNKS modified NUMA at mitosis to regulate spindle assembly and chromatin separation; C: TNKS binds to IRAP and promotes exocytosis of GSVs, catalyzing transmembrane entry of glucose into cells; D: TNKS increases degradation of Axin to reduce stability of degradation complex, thereby increasing level of Wnt signaling.
烟酰胺亚位点在人类PARP家族的成员中保守,许多TNKS抑制剂通过该位点与TNKS结合。XAV939最初是作为PARP1/2抑制剂开发的,其IC50值较弱(对于PARP1和PARP2,分别为2.2 mmol·L-1和0.11 mmol·L-1),后续研究发现,XAV939是TNKS1和TNKS2的更有效的抑制剂(IC50分别为11 nmol·L-1和4 nmol·L-1)[15]。使用XAV939作为先导化合物,Ma等[16]合成了一种名为WXL-8的氨基取代衍生物。通过TNKS1比色酶活性实验,WXL-8表现更有效的TNKS1抑制作用(IC50为9.1nmol·L-1)。WXL-8在HepG2、Huh7、Hep40细胞中抑制细胞增殖和集落形成,通过稳定Axin1和Axin2,降低β-catenin蛋白水平。烟酰胺位点抑制剂通常含有烟酰胺模拟物,其将抑制剂锚定在口袋的底部。模拟烟酰胺的小抑制剂,TIQ-A和菲啶酮的TNKS2的IC50分别为24 nmol·L-1和14 nmol·L-1,PARP1的IC50分别为450 nmol·L-1和300 nmol·L-1,显示出较好的选择性[17]。TIQ-A和菲啶酮的结构非常相似,与TNKS形成的复合物的晶体结构之间没有明显差异。因此,所测量的IC50值的差异可能是由于六元环菲啶酮更有效的疏水相互作用所引起的。Jeffrey等[18]从2-芳基喹唑啉-4-酮化合物开始,发现了吡咯并嘧啶酮化合物AZ6102(4)(Fig 3)。它是一种高效的TNKS1/2抑制剂,选择性是其它PARP家族酶的100倍,在DLD-1细胞中显示5 nmol·L-1的剂量对Wnt通路有明显抑制作用。此外,AZ6102可以在20 mg·L-1静脉溶液中溶解,并在临床前物种中表现出良好的药代动力学性质。Larsson等[19]通过基于片段的配体设计(fragment-based ligand design,FBLD)发现了一系列TNKS2的选择性抑制剂,例如7-(2-氟苯基)-4-甲基喹啉-2(1H)-酮等。通过热转移测定发现的片段靶具有更好的扩展几何矢量,结合模式和XAV939的重叠较少,可以提供更多新颖的化合物。FBLD策略确定了配体结合蛋白中的热点和核心片段。与现有化合物相比,该策略可以产生更有效结合的先导化合物以及新颖的结构框架。吡喃并吡啶酮型抑制剂在各种基于细胞的测定中显示出较高活性,de Vicente等[20]使用基于片段/结构优化策略,发现具有良好吡喃并吡啶酮性能的抑制剂8(Fig 4)。抑制剂8具有适中的全身清除率(28 mg·kg-1·min-1)和分布体积,可被迅速、广泛地吸收。在5mg·kg-1剂量下口服,生物利用度高(49%),以50 mg·kg-1剂量服用时,生物利用度增加至92%。鉴于吡喃并吡啶酮型抑制剂的前景较好,该类抑制剂可用来进行深入的优化和研究。这些小型分子抑制剂结合到PARPs的催化区域,与烟酰胺的结合位点很好地重叠,并显示低纳摩尔效力。与其他结构特异性的PARP相比,TNKSs中的烟酰胺亚位点更具有限制性和疏水性。该位点的疏水性主要是由于位于F环的Pro1034、Phe1035和位于G环的Ile1075。优化后的小分子对TNKS酶具有高选择性,在体外和体内都具有良好的生物利用度和适度的清除率。
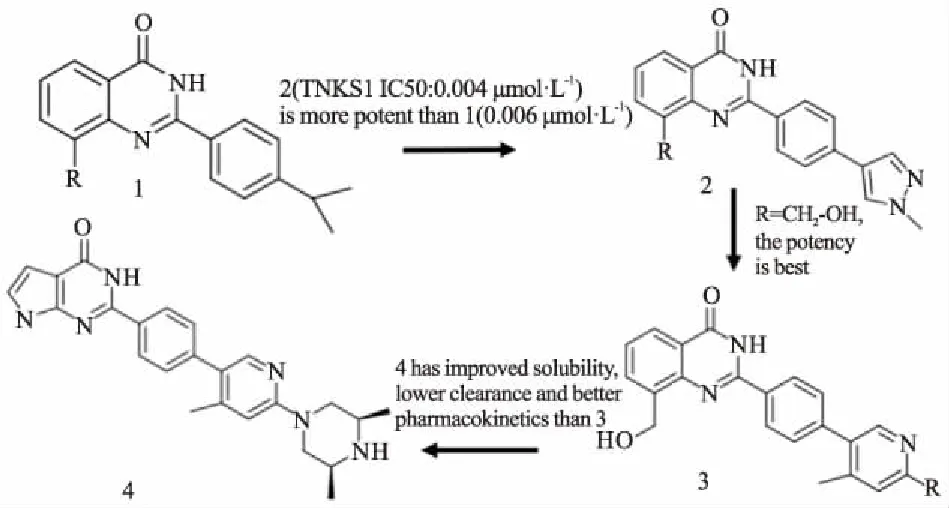
Fig 3 Discovery of AZ6102
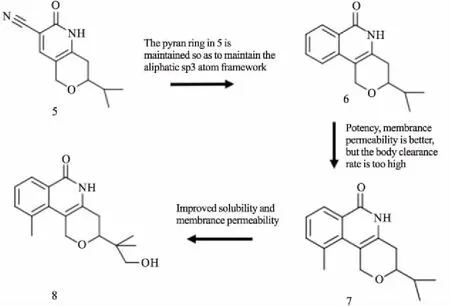
Fig 4 Design and optimization of pyridopyridone inhibitor 8
IWR-1(9)结合到TNKS酶的腺苷亚位点,并引起D环的打开。它的降冰片烯部分结合到活性位点的3个Tyr之间,腺苷部分结合在活性位点α螺旋和D环的1个组氨酸之间[21]。在IWR-1基础上,Bregman等[22]基于结构设计,发现具有高度改善效力和药代动力学性质的化合物12(Fig 5)。二取代的苯并咪唑与IWR1相比,细胞效能明显改善。Huang等[23]在优化上述化合物10期间,通过高通量筛选鉴定化合物13为有效新型TNKS抑制剂。上述两种化合物(10,13)与TNKS1结合的共结晶结构的重叠启发了一个假设,即用2-氨基吡啶代替化合物13的不稳定氨基喹唑啉酰胺(14)可能提高抑制剂在血浆中的稳定性,同时保持与靶蛋白的2个关键的氢键相互作用(Fig 6)。利用高通量筛选,James等[24]发现了新型TNKS抑制剂WIKI4,在所测试的细胞系中,WIKI4均表现出对Wnt信号通路的抑制,包括A375、DLD1结肠直肠癌细胞和人胚胎干细胞。WIKI4对TNKS1/2的IC50值均处于低纳摩尔范围(TNKS1约26 nmol·L-1,TNKS2约15 nmol·L-1)。WIKI4结合模式的结构特征有助于以其作为骨架开发代谢稳定性,药代动力学更优良的新型抑制剂。在人结肠直肠癌细胞中, JW74迅速抑制活性β-catenin,下调Wnt靶基因,包括Axin、SP5。JW74衍生物G007-LK是一种有效的TNKS抑制剂,其优于多种其他同工酶的选择性,对测试的激酶、磷酸酶和GPCRs没有抑制作用,并且在体外具有良好的活性[25]。G007-LK在Ⅰ期临床试验中代谢稳定,并且显示出优异的药代动力学特征。最近,Okada-Iwasaki等[26]使用转录报告基因系统在突变型结肠癌DLD-1细胞中进行高通量筛选,发现了选择性Wnt通路抑制剂K-756。K-756通过抑制Wnt /β-catenin途径,抑制APC突变型结肠直肠癌COLO 320DM和SW403细胞的生长。虽然ARTD1/2抑制剂在BRCA突变的治疗中被证明是有效的,其副作用如诱发基因组不稳定性可能是潜在隐患。因此,与较少选择性的TNKS抑制剂如XAV939相比,K-756可能是抗肿瘤治疗药物开发的更好候选者。可口服给药的选择性化合物K-756可以作为进一步开发更有效的衍生物的先导化合物。尽管与腺苷亚位点结合的化合物发现方法不同,它们都具有2个保守的氢键受体,与D环的组氨酸和各种疏水区的π-π堆积。通常在TNKS选择性腺苷位点和烟酰胺位点结合物之间共享1或2个疏水作用。到目前为止,仅与腺苷亚位点结合的抑制剂没有被证实可作用于其他PARPs,提示这些化合物的高度选择性。
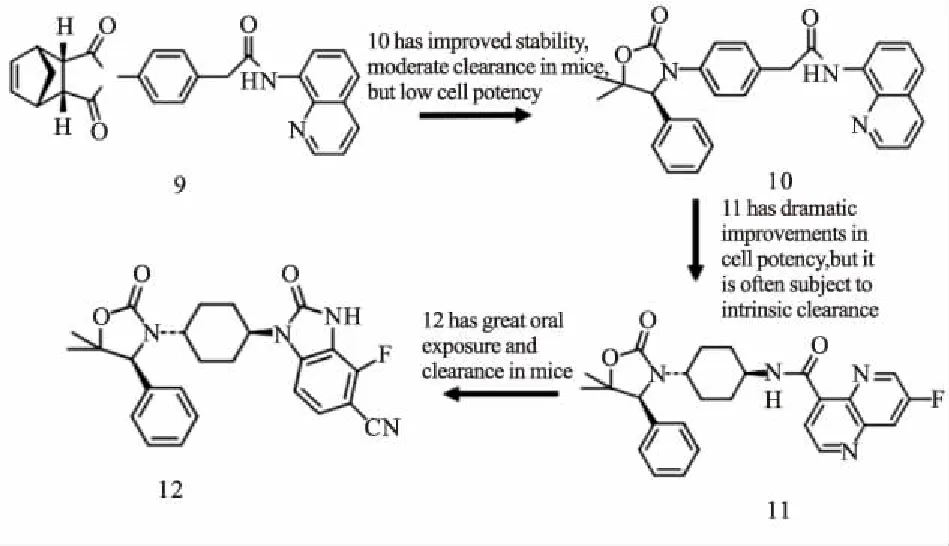
Fig 5 Design and optimization of oxazolidinone inhibitor 12
烟酰胺亚位点与腺苷亚位点的发现,促进了研究者去设计同时阻断这2个位点的抑制剂。EB-47被证实是双位点抑制剂[17],作为NAD+模拟物,EB-47的结合类似于NAD+的结合。在TNKS2中,它在烟酰胺亚位点处发生常规的相互作用。在腺苷亚位点,化合物的核糖羟基与His1031和Ser1033(它们在PARP家族中保守,但不被其他抑制剂所利用)相互作用(Fig 7)。使用以IWR-2药效基团为基础的亚结构检索模型,Bregman等[27]确定了1种对TNKS1具有低纳摩尔效力的长喹唑啉酮化合物。化合物在细胞实验中具有比IWR-1和XAV939更强的Wnt通路抑制作用(TNKS1 IC50:0.008 μmol·L-1,TNKS2:0.002 μmol·L-1)。近期,诺华公司发布的双位点抑制剂NVP-TNKS656在小鼠实验结果,显示具有良好的药动学特性、生物利用度和改善的选择性(TNKS2特异性抑制剂)[28],使其成为化学骨架良好的候选者。
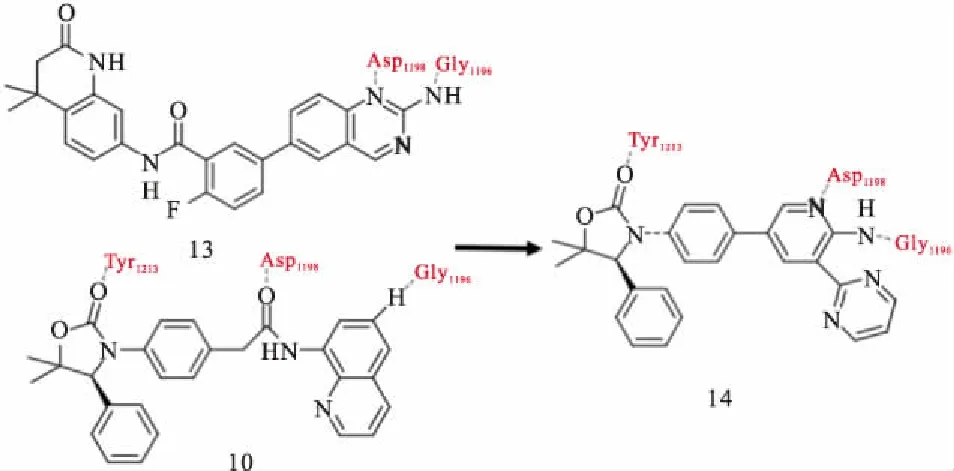
Fig 6 Design and optimization of oxazolidinone type 14
Fig 7 The chemical structure of EB-47(A) and crystal structure
4 展望
TNKS是富有挑战性的药物靶点,它们在个体细胞、健康或疾病中发挥的功能还需要更加深入的理解。TNKS抑制剂是一个相对较新的研究领域,这个未知的领域对于药物发现研究人员来说有很大的潜力。选择性是阐明TNKS酶在开发下一代TNKS抑制剂作为治疗剂发挥功能的重要方面。随着TNKS抑制剂的分子机制和结合模式的日益了解,将推动下一代针对PARPs家族成员量身定制的多靶点活性的先导化合物的设计。例如了解特定的TNKS-TNKS抑制剂相互作用的详细结构信息、结合口袋周围的化学空间,以及具有能量可接近的几何形状的整体形状,有助于发现更精细的、更高效力和改善的选择性下一代TNKS抑制剂。多学科的协调能更好地帮助优化先导化合物。此外,多样性定向合成(DOS)、动态组合化学(DCC)和HTS技术(例如微阵列)的结合,可以用于探索与生物相关的化学空间的TNKS,并进一步彻底改变先导化合物的发现和优化步骤。目前,所有TNKS抑制剂靶向供体NAD+结合位点,另外两种可药用的位点存在于受体位点和锚蛋白重复序列。在使用自动修饰作为信号的体外酶活性筛选中,没有发现锚蛋白重复抑制剂,蛋白质与蛋白质相互作用目前也较难以用抑制剂靶向。然而,锚蛋白重复序列和受体位点提供用于选择性靶向TNKS酶的新的场所,因为锚定蛋白重复不存在于其它PARP中,大大提高了抑制剂的选择特性。另外,TNKS的抑制似乎具有强烈的微环境依赖性,TNKS抑制的生物学结果将受其他细胞途径活性影响,例如细胞NAD+水平、缺氧条件和其他代谢改变。TNKS和PARP1/2双重抑制剂仍然是新型抗癌药物开发的新策略。抑制TNKS似乎在治疗上很有吸引力,但仍需要药理学改进的化合物或与其他疗法的组合以发挥抗癌作用。因此,TNKS抑制剂可能需与其他的细胞途径抑制剂合用来提高治疗效果。特别是在癌症背景下,几种PARP酶的抑制剂鸡尾酒疗法可能是更有效的策略。此外,与其他PARP同工酶选择性抑制剂联用也是未来的探索方向。开发更有效和更具特异性的TNKS酶抑制剂不仅能够使我们进一步深入认识TNKS的功能,更重要的是推动肿瘤治疗相关的临床研究。
猜你喜欢
检察风云(2022年5期)2022-04-05
家庭医药(2021年13期)2021-12-03
中医眼耳鼻喉杂志(2021年2期)2021-07-21
科技创新与应用(2021年16期)2021-01-04
中国化妆品(2020年2期)2020-04-13
第一财经(2019年8期)2019-08-26
中国化妆品(2019年1期)2019-04-18
中南民族大学学报(自然科学版)(2019年1期)2019-04-04
中国医药指南(2017年3期)2017-11-13
中国洗涤用品工业(2017年6期)2017-08-22
