
激活Sonic hedgehog通路改善缺血缺氧心肌细胞DNA损伤
2018-09-08 03:05梁关凤李素娟邱晓霞张贵平罗健东袁文常
中国药理学通报 2018年9期
梁关凤,李素娟,邱晓霞,张贵平,罗健东,袁文常,侯 宁
(1. 广州医科大学药学院药理学教研室,广东 广州 511436;2.广东省第二人民医院药学部,广东 广州 510000;3. 广州医科大学附属第五医院检验科,广东 广州 510700)
急性心肌梗死(acute myocardial infarction,AMI)是指因持久而严重的心肌急性缺血、缺氧所引起的部分心肌坏死。AMI后,心肌细胞严重损伤和丧失,直接导致心脏舒张和收缩功能下降、心室重构等。因此,提高AMI心肌细胞存活率有重要意义。研究表明,Sonic hedgehog(Shh)通路在AMI心肌细胞中被活化,该通路激活后,心肌细胞的存活率增加[1]。然而,此通路在急性心梗时对心肌细胞自身修复的调控机制尚不明确。近年来,DNA损伤在心血管疾病中的作用日益引起关注,在细胞进行生命活动时,DNA会不可避免地遭受各种内源和外源损伤,氧化应激、代谢紊乱、缺血缺氧等是其主要的诱发因素[2]。目前认为,细胞在受到DNA损伤后,会在损伤部位募集DNA损伤反应蛋白和DNA修复相关蛋白,启动DNA损伤修复[3]。
心肌梗死后,血循环中游离DNA 水平明显升高[4]。在动脉粥样硬化病人的斑块组织内,DNA损伤反应蛋白-毛细血管扩张症突复基因( ataxia telangiectasia mutated gene,ATM) 磷酸化水平升高[5]。在肾脏缺血/再灌注模型中,存在组蛋白(histone,H2AX)、ATM等DNA损伤相关蛋白磷酸化水平增高[6]。不同形式的DNA 损伤能够被不同蛋白复合物识别,通过与磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)家族成员ATM、ATM和Rad3相关蛋白(ATM and Rad3-related protein,ATR)、DNA依赖性蛋白激酶催化亚单位(DNA-dependent protein kinase catalytic subunit,DNA-PKcs)等结合,启动 DNA 损伤反应及其后续修复途径[7]。那么,在梗死的心肌组织中,心肌细胞的急性缺血缺氧是否导致DNA损伤的发生?Shh通路与心肌细胞的DNA损伤之间又有着怎样的联系?为回答上述问题,本研究建立Sprague Dawley(SD)乳鼠体外心肌缺氧模型,通过检测 DNA损伤反应通路中关键蛋白表达,验证 DNA损伤是否参与心肌缺氧性损伤;同时应用Shh通路的激动剂SAG1.3和抑制剂GANT61来调控该通路,观察其对心肌细胞DNA损伤的影响。
1 材料与方法
1.1材料
1.1.1实验动物 新生1~3 d的SD乳鼠,雌雄不限,购自广东省实验动物中心。
1.1.2试剂 DMEM干粉、L-15细胞培养液、胎牛血清,购于美国Gibco公司;Shh通路激动剂SAG1.3、抗p-ATM、γH2AX一抗,购自Merck公司;Shh通路抑制剂GANT61购自MCE公司;p-ATR、p53结合蛋白1(p53-binding protein 1,53BP1)、GAPDH、Bax、Bcl-2、p53、p-p53一抗,购自Cell Signaling Technology公司;Gli1一抗购自R&D公司;Gli2一抗购自Proteintech公司。
1.1.3仪器 低温高速离心机(德国Hettich公司);垂直电泳槽、转膜仪(美国Bio-Rad公司);荧光正置显微镜(日本Nikon公司);超净工作台(苏州仪器厂);凝胶成像分析系统(美国UVP);自动洗片机(美国Kodak)。
1.2乳鼠心肌细胞分离培养新生1~3 d SD大鼠用75%乙醇清洁1~2次,在超净工作台无菌条件下,用眼科剪沿左侧肋骨快速打开乳鼠的胸腔暴露搏动心脏,弯镊取出左心室心肌组织,放入装有30 mL HBSS离心管中,洗3遍直至洗净残留血液。洗干净的心肌组织移至100 mm培养皿中,修剪后剪碎成0.2 mm的小块,加入新鲜的8 mL HBSS和2 mL 0.25%的胰酶,4℃冰箱消化13 h。次日,加入2 mL的FBS终止消化后,加入Ⅱ型胶原酶,终浓度为 0.1%,于37℃水浴摇动45 min,取出加入L-15细胞培养液吹洗10次过滤,于室温中静置20 min,离心,加入含10% FBS的DMEM吹打成细胞悬液,种于100 mm培养皿中,放在正常细胞培养箱中45 min进行差速贴壁。取出培养皿,收集液体离心,弃上清留沉淀,加入含15% FBS的DMEM。在显微镜下计数,接种细胞,24 h后换液,可见细胞搏动大于95%即可进行后续实验。
1.3细胞糖氧剥夺(deprivationofoxygenandglucose,OGD)模型的建立心肌细胞分离培养24 h后,换成无EBSS,分别加入Shh信号通路激动剂SAG1.3、抑制剂GANT61,将细胞放置于37℃无氧(94% N2、5% CO2、1% O2)培养箱培养,进行以下实验。
1.4实验分组(1)时间点观察分为,Control组:空白对照组;OGD组:氧葡萄糖剥夺处理组,细胞只给予无糖EBSS培养基处理,缺氧培养至预定时间(3、6、12、24 h);(2)药物处理分组:Control组:空白对照组;OGD 6 h、OGD 12 h:氧葡萄糖剥夺处理组,细胞只给予无糖EBSS培养基处理,缺氧培养至6 h或12 h;OGD 6 h+SAG1.3、OGD 12 h+SAG1.3:氧葡萄糖剥夺处理+SAG1.3组,无糖EBSS培养基处理细胞,缺氧培养至6 h或12 h;OGD 6 h+GANT61、OGD 12 h+ GANT61:氧葡萄糖剥夺处理+GANT61组,无糖EBSS培养基处理细胞,缺氧培养至6 h或12 h。
1.5Westernblot检测取等量的各组蛋白上样,电泳,分子质量低于200 ku的蛋白使用SDS-PAGE凝胶,在100 V恒压下电泳,250 mA恒流转膜;分子质量大于200 ku的蛋白,使用NuPAGETM3%~8% Tris-Acetate Gel在150 V恒压电泳1.5 h,30 V恒压转膜3 h。转膜结束后,取出膜,用5%脱脂奶粉室温封闭PVDF膜1 h;一抗4℃孵育过夜,TBST洗膜3次,每次10 min;二抗室温孵育1 h,TBST洗膜5次,每次10 min;取出膜,增强化学荧光发光试剂中反应1 min;曝光成像后进行扫描。
1.6细胞免疫荧光法按照1×108·L-1的密度,将乳鼠心肌细胞接种到6孔板中(6孔板中预先在底部放置1片已消毒的玻璃片,并用0.1%明胶预处理)。处理细胞后,倒去培养基,用PBS清洗3次,加入4%多聚甲醛固定10 min,再用PBS清洗3次,-20℃保存孔板。取出细胞爬片,裁成1 cm2大小放置在载破片上,滴加新鲜PBS润洗5 min。将载玻片浸入1% Triton 100中,室温放置30 min,然后用PBS清洗,5 min×3次;将载玻片浸入100 mmol·L-1甘氨酸,室温放置20 min,用PBS清洗,5 min×3次;10%山羊血清封闭1 h后,加入一抗,4℃孵育过夜,PBS-T漂洗4次,荧光二抗室温避光孵育45 min,PBS-T漂洗后,加入DAPI室温孵育10 min,ddH2O2清洗后封片,荧光显微镜下分析样本,拍照。
1.7MTT法检测细胞存活率按照1×104个/孔的密度,将乳鼠心肌细胞接种到96孔板中,加入含15% FBS的DMEM培养基,每孔100 μL;于37℃、5% CO2细胞培养箱中培养24 h后,用PBS清洗细胞2次,含10% FBS的DMEM培养基换液;加实验处理因素处理细胞;每孔加入MTT 50 μL,细胞培养箱中孵育4 h;小心吸出上清液,每孔加入100 μL DMSO,轻轻振荡15 s,混匀;在酶标仪570 nm波长处检测吸光度值。

2 结果
2.1心肌细胞OGD各时间点Shh通路标志蛋白及DNA损伤通路蛋白的表达Shh通路活化后,其下游蛋白Gli1/2表达明显上调,因此,本实验以Gli1/2表达为Shh通路活化的标志物。Fig 1A显示,OGD时Gli1/2表达随OGD时间延长逐渐增高,6 h时表达最高,其后逐步下降。组蛋白H2AX第139位丝氨酸残基磷酸化形成的γH2AX是DNA损伤检测的重要标志物。如Fig 1B所示,OGD时可诱导心肌细胞γH2AX表达升高,随OGD时间延长,γH2AX的表达在6 h时达到高峰,随后其表达逐步降低。DNA损伤反应蛋白p-ATM的表达变化与γH2AX表达一致;另一DNA损伤反应蛋白p-ATR的表达与γH2AX相反,在心肌细胞OGD 12、24 h升高。进一步检测凋亡相关蛋白和凋亡标志物发现,随着OGD时间延长,心肌细胞中凋亡标志蛋白p53,cleaved-caspase-3表达逐渐增加,Bcl-2/Bax比例逐渐降低(Fig 1C)。提示OGD诱导的心肌细胞缺血缺氧通过促进DNA损伤诱导凋亡。
2.2心肌细胞OGD各时间点γH2AX的表达如Fig 2所示,心肌细胞随OGD时间延长,核内γH2AX表达逐步增高,γH2AX在OGD 6 h时表达最高,之后逐步下降,该结果与Western blot结果一致。
2.3OGD6h调控Shh通路对心肌细胞DNA双链断裂(doublestrandbreak,DSB)损伤的影响Fig 3结果显示,Shh通路的激动剂SAG1.3、抑制剂GANT61能特异性调节心肌细胞Gli1/2表达。OGD 6 h时,心肌细胞Gli1/2表达升高,DNA损伤标志物γH2AX、损伤反应蛋白p-ATM表达同步升高,另一损伤反应蛋白p-ATR未见明显变化。与OGD组比较,给予Shh通路抑制剂GANT61时,Gli1/2减少,γH2AX、p-ATM表达增多,p-ATR无明显变化;给予Shh通路激动剂SAG1.3时,Gli1/2增多,γH2AX、p-ATM表达减少,p-ATR无明显变化。
2.4OGD6h调控Shh通路对DSB损伤的影响Fig 4免疫荧光结果显示,OGD 6 h,与OGD组相比,Shh通路抑制剂GANT61诱导γH2AX表达上调;Shh通路激动剂SAG1.3明显抑制γH2AX表达,该结果与Western blot结果一致。
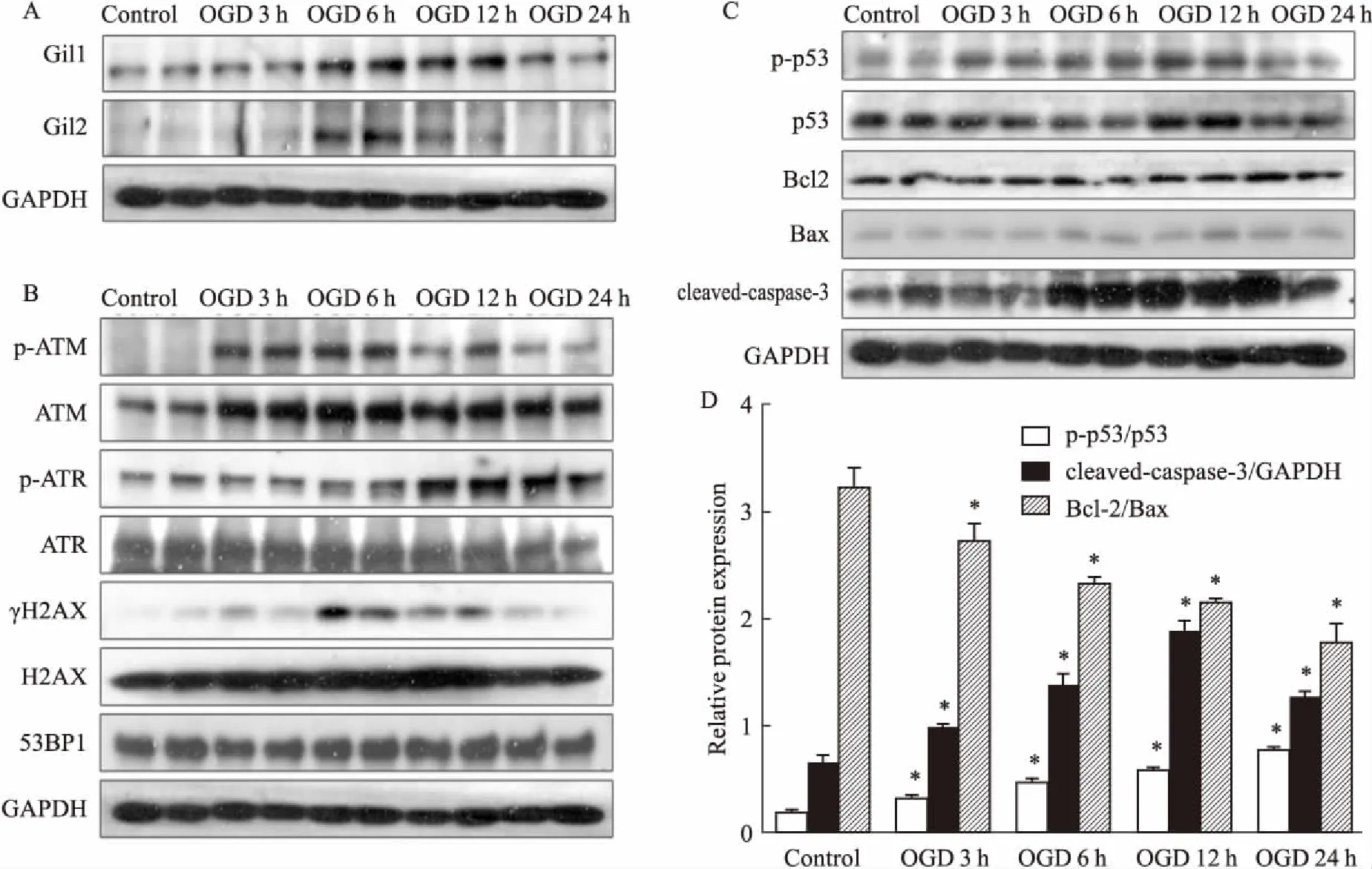
Fig 1 Expression of Shh pathway proteins (A), DNA damage proteins(B), and apoptotic related proteins(C)
*P<0.05vscontrol
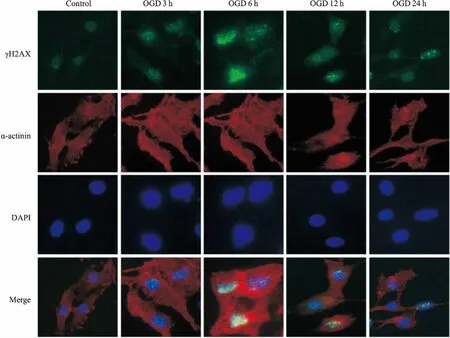
Fig 2 Expression of γH2AX at different time points in neonatal rat cardiomyocytes deprivedof glucose and oxygen detected by immunofluorescence(×400)
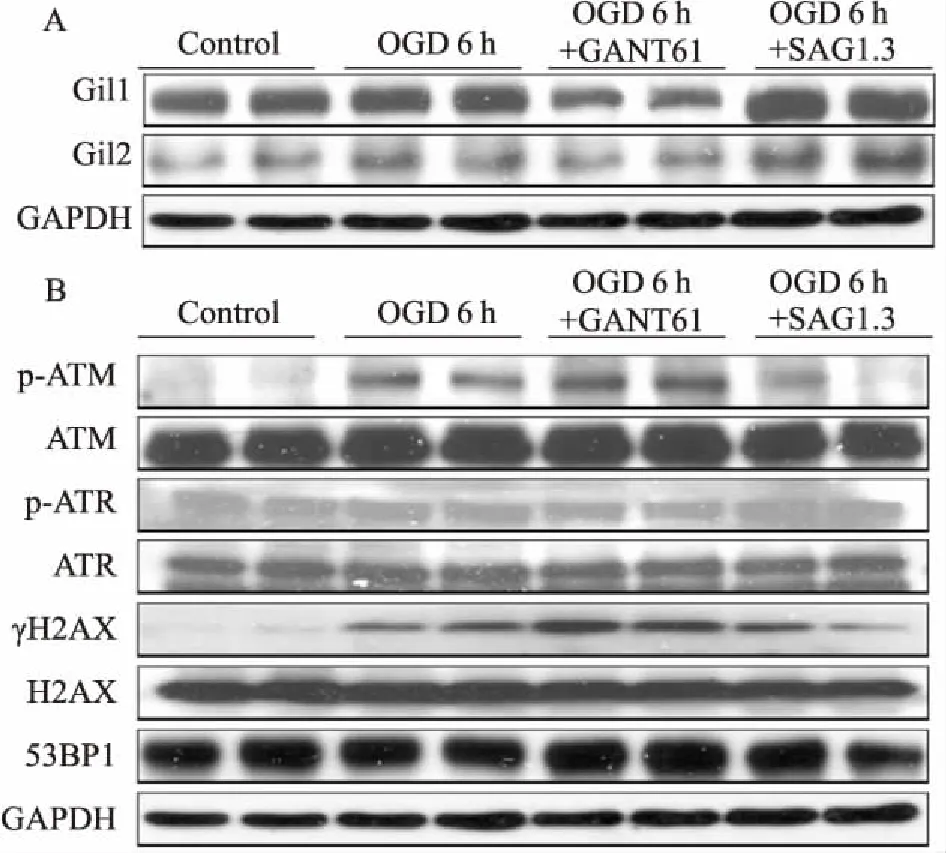
Fig 3 Expression of Gli1/2(A) and DNA damage proteins (B) at OGD 6 h detected by Western blot
2.5OGD12h调控Shh通路对DSB损伤的影响如Fig 5所示,与OGD 6 h的实验结果一致,OGD 12 h给予Shh通路抑制剂GANT61后,Gli1/2减少,γH2AX、p-ATM表达增多,p-ATR略有升高;给予Shh通路激动剂SAG1.3时,Gli1/2增多, γH2AX、p-ATM表达减少,p-ATR表达经与ATR条带灰度值比较,未见明显变化。
2.6OGD12h调控Shh通路对DSB损伤的影响Fig 6免疫荧光结果显示,OGD 12 h时,与OGD组相比,Shh通路抑制剂GANT61诱导γH2AX表达增多;Shh通路激动剂SAG1.3明显抑制γH2AX表达。该结果与Western blot结果一致。
2.7OGD6、12h调控Shh通路对心肌细胞凋亡的影响Tab 1、Fig 7结果显示,给予Shh激动剂SAG1.3处理后,OGD 6、12 h时,心肌细胞的存活 率均明显提高,细胞凋亡标志蛋白p-p53、cleaved-caspase-3表达减少、Bcl-2/Bax比值增高,细胞凋亡减少。与之相反,经Shh抑制剂GANT61处理的心肌细胞,OGD 6、12 h细胞存活率降低,p-p53、cleaved-caspase-3表达上调,Bcl-2/Bax比值降低,凋亡细胞数目明显增多。


Group6 h12 hControl0.569±0.1400.569±0.150OGD0.294±0.058**0.336±0.025**OGD+GANT610.231±0.021#0.247±0.012#OGD+SAG1.30.306±0.042#0.352±0.036#
**P<0.01vscontrol;#P<0.05vsOGD

Fig 4 Expression of γH2AX when Shh pathway was regulated at OGD 6 h detected by immunofluorescence(×400)
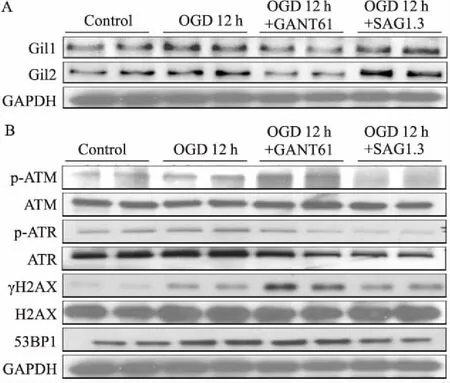
Fig 5 Expression of Gli1/2 (A) and DNA damage proteins (B) at OGD 12 h detected by Western blot
3 讨论
AMI是威胁人类健康的常见心血管疾病之一,研究AMI后心脏功能改善和心肌修复的具体机制对临床治疗具有重要的现实意义。Shh是Hedgehog家族中分布最广、研究最多、功能最重要的一类蛋白。大量研究表明,分子质量为19 ku 的Shh与其受体Ptch1结合,促进核转录因子 Gli1、Gli2入核,激活Hedgehog信号通路,通过促进细胞的增殖、干细胞的存活、血管生成等,对胰腺、骨髓、心血管系统等组织损伤的修复发挥重要的调节作用[8]。大量研究表明,AMI心脏中Shh信号通路是被激活的,该通路活化后可促进心肌梗死区域及梗死周边区域毛细血管新生,改善心脏功能,缩小梗死面积。但其对心肌细胞自身修复能力的调控作用及机制尚不完全清楚。
研究发现,多种因素导致的DNA损伤中,γH2AX即组蛋白H2AX第139位丝氨酸磷酸化蛋白表达均增高;在DNA损伤修复反应早期,H2AX的磷酸化是最早发生的分子事件之一,因此,γH2AX已经被广泛认定为DNA损伤的分子标志物。γH2AX可作为起始信号分子,将下游相关蛋白募集到损伤位点,进而启动细胞的周期阻滞、修复、凋亡等[9]。H2AX的磷酸化主要受到PI3K家族成员ATM、ATR、DNA-PKcs等蛋白的催化调控。大量研究发现,不同环境因素诱导的DNA损伤通过活化不同的PI3K成员,诱导H2AX磷酸化,从而启动DNA损伤修复应答系统。由DNA复制差的压力应激反应诱导的H2AX磷酸化主要依靠ATR催化,与ATM、DNA-Pks无关;而在电离辐射诱导的细胞内,γH2AX表达主要是由p-ATM的自磷酸化催化[10]。我们前期在研究氧化应激诱导的心肌细胞DNA损伤时也发现,H2O2诱导的H2AX磷酸化以及γH2AX簇集点的形成主要是通过ATM的磷酸化完成的[11]。
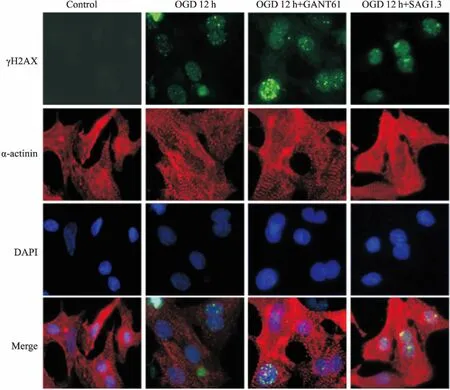
Fig 6 Immunofluorescence detected the expression of γH2AX when Shh pathway was regulated at OGD 12 h(×400)

Fig 7 The expression of apoptotic proteins and the cardiomyocytes survival rate whenregulate the Shh pathway at OGD 6 h (A) and OGD 12 h
本研究以原代培养的乳鼠心肌细胞建立OGD的体外缺血缺氧模型,通过Western blot检测DNA损伤标志性蛋白γH2AX,以及DNA损伤的关键应答蛋白ATM、ATR的表达。结果显示,当心肌细胞缺氧时,γH2AX表达明显增高,在OGD 6 h时表达达高峰,此时较多的心肌细胞已经出现较大损伤,随后γH2AX的表达逐步下降,心肌细胞凋亡增加。ATM磷酸化表达趋势与γH2AX一致,说明缺血缺氧可通过诱导心肌细胞ATM磷酸化,促进H2AX磷酸化,随后启动DNA损伤修复反应。
本项目组前期的体内外研究中已经明确,Shh通路活化在心肌梗死、氧化应激诱导的心肌细胞凋亡、自噬中发挥重要保护作用[12-13]。已有的研究显示,活化的Shh对离子辐射造成的人肝癌细胞DSB具有保护作用[14];抑制人结肠癌细胞Gli表达,可直接激活DSB反应激酶,诱导核内γ-H2AX表达增多,活化凋亡诱导蛋白,导致癌细胞广泛凋亡[15]。但是,目前该通路对心肌DNA损伤的调控关系尚不清楚。本研究进一步探讨Shh通路与缺氧诱导的心肌细胞DNA损伤的关系。实验结果显示,在OGD后,心肌细胞Gli1/2表达上调,Shh通路被激活;Gli1/2在OGD 6 h也出现与γH2AX一致的表达高峰,提示Shh信号通路可能参与心肌细胞DNA损伤的应答过程。随后分别使用Shh通路激动剂SAG1.3、抑制剂GANT61调控心肌细胞Shh通路的活性,结果发现,当给予Shh通路激动剂SAG1.3时,γH2AX、p-ATM表达明显减少,细胞凋亡标志物表达减少,细胞生存率提高;而抑制剂GANT61则加重DNA损伤,γH2AX、p-ATM表达明显增加,细胞凋亡标志物表达增多。由此可见,Shh信号通路可通过抑制p-ATM/γH2AX通路,减轻DNA损伤,从而在缺血缺氧诱导的心肌细胞凋亡中发挥重要保护作用。但Shh通路如何影响ATM的磷酸化水平,其具体的调控机制尚需深入探讨。本研究首次在体外研究中证实Shh通路与心肌细胞DNA损伤的关系,后续将在心肌梗死动物模型中深入研究Shh通路对心肌DNA损伤和修复的调控作用及机制。
综上所述, Shh通路活化可通过影响DNA损伤修复通路,抑制心肌缺血缺氧诱导的细胞凋亡,本研究为改善AMI细胞凋亡、促进心肌修复提供新的线索和思路。
(致谢:本实验在广州医科大学动物实验中心、药理实验室及基础医学实验教学中心机能实验室完成,在此对实验过程中给予指导和帮助的老师表示感谢!)
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
波谱学杂志(2022年1期)2022-03-15
世界科学技术-中医药现代化(2021年7期)2021-11-04
食品安全导刊(2021年20期)2021-08-30
中华养生保健(2020年9期)2021-01-18
自我保健(2019年10期)2019-12-11
中国实验动物学报(2019年4期)2019-09-03
天津医科大学学报(2019年6期)2019-08-13
分析化学(2017年12期)2017-12-25
中成药(2017年8期)2017-11-22
