
妊娠中期羊水过少胎儿的遗传学分析
2018-09-07 13:10:26李晓青王利群刘晋红游艳琴周红辉张蔓丽张鑫悦卢彦平
解放军医学院学报 2018年8期
李晓青,王利群,刘晋红,游艳琴,周红辉,张蔓丽,张鑫悦,卢彦平
解放军总医院,北京 100853 1妇产科;2病理科
妊娠中期以后羊水的主要来源为胎儿尿液,羊水过少主要依靠超声检查发现,诊断标准是最大羊水暗区垂直深度(maximum vertical pocket,MVP)≤2 cm。此时应着重关注胎儿是否存在畸形可能,如肾缺如、肾发育不全、输尿管或尿道狭窄、膀胱出口梗阻、多囊性肾病、Meckel综合征等,以上畸形均可能导致尿液产生过少或排出受阻,最终表现为羊水过少。而其中常染色体隐性遗传病是重要的原因之一[1]。随着高通量测序技术的发展,染色体微阵列分析(Chromosomal microarray analysis,CMA)及全外显子组检测技术(Whole exome sequencing,WES)在遗传病诊断中具有很大的优势[2-3]。2013年美国妇产科医师学会(American College of Obstetricians and Gynecologists,ACOG)和美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics,ACMG)提 出 了基因芯片在产前诊断领域的应用规范[4-5],尤其是对一些临床表型不典型、外显率下降或多变表型遗传性疾病,这些疾病容易给临床医生带来误导,使用靶向基因的panel容易产生阴性结果。而从技术角度看,WES为编码变异提供全基因组范围筛查平台,Blue等[6]在家族性先天性心脏病的研究中证明了使用WES能更容易解决这一问题。但是WES也有其局限性,WES并不能检测所有的潜在因果关系的基因突变型,并可能出现严重遗漏,如病理性重复扩增和大多数CNVs[7]。由于本研究中的病例均为羊水过少病例,传统核型分析存在困难,采用对于染色体病筛查更全面的SNP-array平台可以同时对染色体微缺失、微重复进行筛查。同时采用WES技术进行单基因病的筛查,两种检测手段共同应用探究妊娠中期羊水过少患者的遗传学病因。
对象和方法
1 研究对象 2015年9月- 2017年12月我院收治的妊娠中期羊水过少病例15例,所有病例于两家其他医院各行1次超声检查或于我院间隔1 ~ 2周行2次以上的超声重复检查确诊,并引产。纳入标准:1)核对孕周在妊娠14 ~ 27+6周;2)超声提示羊水MVP≤2 cm;或妊娠14 ~ 27+6周发生胎死宫内,同时超声提示羊水过少。排除标准:1)胎膜早破;2)孕期有毒物接触史或有血管紧张素转换酶抑制剂(angiotensin-converting enzyme inhibitors,ACEI)或血管紧张素Ⅱ受体阻滞剂(angiotensinⅡreceptor blocker,ARB)类降压药、前列腺素合成酶抑制剂用药史;3)孕期有感染史;4)多胎妊娠。所有家庭签署知情同意书,均经院伦理委员会审批。15例羊水过少引产(胎死宫内)病例临床资料见表1。
2 研究方法 取胎儿大腿内侧皮肤组织1 cm2,委托贝康公司进行基因芯片检测,使用human cyto-12芯片和扩增、杂交试剂盒(Illumina),参照Infinium HD Assay标准操作流程进行扩增、杂交、扫描和分析,结果判读参照DGV、CHOP database和OMIM数据库。智因东方公司完成全外显子检测,外显子区的捕获采用罗氏NimbleGen 公司的Seq EZ Exome Enrichment Kit V2.0捕获探针进行人全外显子组捕获,覆盖人基因组中19 119个基因的编码区,总共捕获区间大小是40 Mb。测序平台使用Illumina公司的HiSeq X测序仪,测序策略为PE150,平均测序深度为100×,测序数据量≥8 G,测序质量(Q20)≥90%,测序质量(Q30)≥85%。
结 果
1 胎儿CMA检测 15例均未发现异常。
2 胎儿WES检测 12例中有10例未发现可疑致病基因,2例发现可疑致病基因:1例ACE基因c.1631T>C(p.L544P)和c.3070-3071delCT(p.L1024Lfs*17)复合杂合突变,相关疾病为肾小管发育不良 (renal tubular dysgenesis,RTD);1例ANKS6 c.2394+1G > A(IVS13)、c.621(exon2)_c.622(exon2)insTGGTG复合杂合突变,相关疾病为肾消耗病16型 (nephronophthisis-16,NPHP16)。
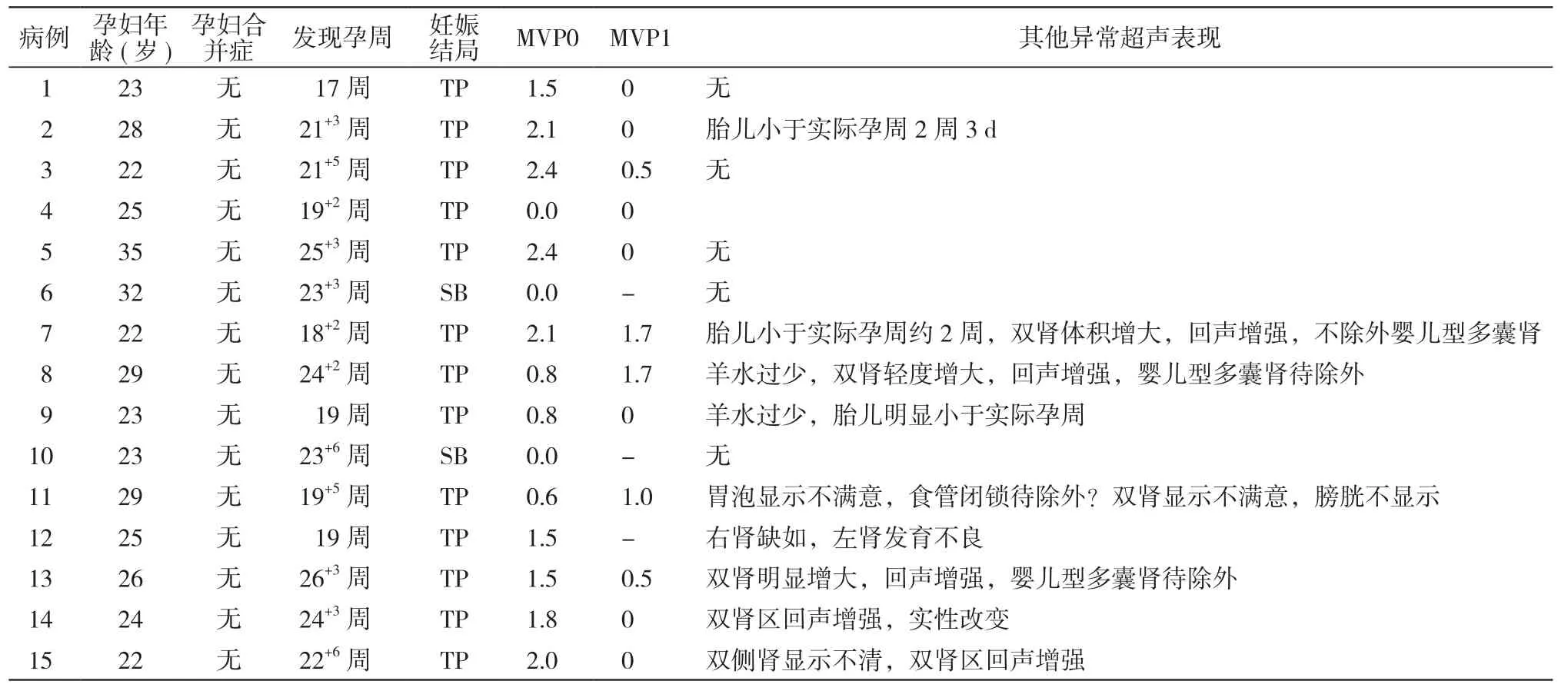
表1 15例孕中期羊水过少患者临床资料Tab. 1 Clinical data about 15 cases with oligohydramnios in the second trimester
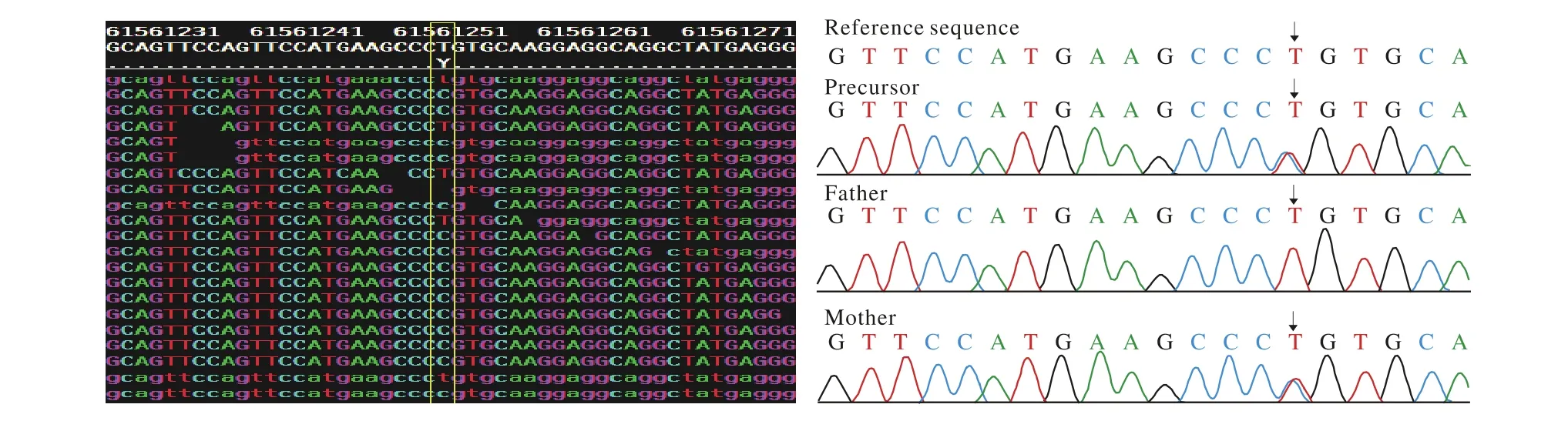
图1 ACE基因突变位点c.1631T>C (dbSNP数据库未收录),该突变染色体位置为chr17:61561254,该突变来源于先证者之母,导致氨基酸改变为p.544,L>P,经蛋白结构预测结果为有害Fig. 1 ACE gene mutation c.1631T>C is located at chr17: 61561254, which is not included in dbSNP database. The mutation comes from the mother of the proband, resulting in amino acid alteration of p.544, L>P. The result of protein structure prediction is harmful

图2 ACE基因c.3070-3071delCT (dbSNP数据库未收录),该突变染色体位置为chr17:61561254,该突变为移码突变,来源于先证者之父,导致氨基酸改变为p.L1024Lfs17,该突变为移码突变,理论上序列与原来的完全不同,对所编码蛋白质的功能影响比较明显Fig. 2 ACE gene c.3070-3071delCT is located at chr17:61561254, which is a frameshift mutation and it is not included in dbSNP database. It originates from the father of proband and causes amino acid change to p.L1024Lfs17. The frameshift mutation is theoretically different from the original one, and its effect on the function of the encoded protein is signif i cant

3 2例存在可疑致病基因突变病例的临床资料ACE基因突变先证者父母连续两次于妊娠24周左右发现羊水过少,最终引产。2011年第1次妊娠,引产后未行进一步检查。2013年第2次妊娠,24周产前超声提示羊水过少,最大平面深0.9 cm,胎儿双肾增大,实质回声增强,皮髓质分界不清,未提示其他胎儿发育异常,观察至孕29+1周仍羊水过少,于外院行引产术。引产后胎儿行尸检,胎儿胸腹腔内各脏器未见明显畸形。为进一步明确致病原因,夫妻双方染色体核型分析,结果均正常,夫妻双方无相关遗传家族史,且双方肾超声检查均正常。胎儿行SNP-array检测,未发现变异。2015年取得胎儿DNA,并留取夫妻全血行全基因组外显子检测,结果先证者突变基因为ACE,突变位点为c.1631T>C(图1)和c.3070-3071delCT(图2)复合杂合突变。
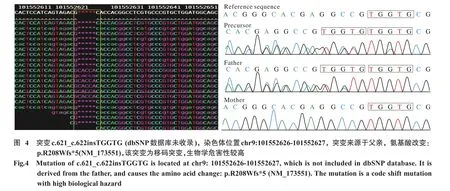
ANKS6基因突变先证者的父母孕26+3周于外院超声检查发现羊水过少,最大羊水池深度1.5 cm,胎儿双肾增大就诊于我院,孕27周+5 d于我院行产前诊断超声:羊水过少(MVP=0.5 cm),胎儿双肾明显增大,回声增强,考虑婴儿型多囊肾待除外。胎儿尸检外观未见明显畸形,脏器解剖发现双肾明显增大,肾剖面双肾成颗粒状改变,镜下病理双肾均见大小不等的囊泡,肾皮质层较薄,肾小球及肾小管数量均较少,肾小管和肾小球形态异常(图3)。取胎儿组织行WES检测结果提示先证者为ANKS6基因c.621(exon2)_c.622 (exon2)insTGGTG(图4)和c.2394+1G>A(IVS13)复合杂合突变(图5)。
讨 论
近年来,高通量测序技术逐步应用于遗传病基因检测[8-13],为疑难病例的诊断带来希望。本研究中,在充分排除了母体、药物、胎盘、脐带等因素后,采用基因芯片技术及WES技术,15家系均未发现染色体结构畸变,WES技术一次性对全外显子组进行序列分析,为2家系寻找到了可疑致病基因:1例ACE基因c.1631T>C(p.L544P)和c.3070-3071delCT(p.L1024Lfs*17)复合杂合突变,涉及疾病为肾小管发育不良;1例ANKS6c.2394+1G > A(IVS13)、c.621(exon2)_c.622(exon2)insTGGTG复合杂合突变,涉及疾病为肾消耗病16型。以下分别对发现可疑致病基因的2家系进行病例分析。

RTD家系临床表现和基因型分析符合遗传学共分离现象。我们通过蛋白质结构及功能软件预测,以上变异会导致ACE基因编码的蛋白质发生改变,c.3070-3071delCT为框移突变,引起氨基酸改变的为p.L1024Lfs*17即第20外显子第3 070 ~3 071位缺失碱基CT,导致编码的蛋白从第1 064位氨基酸开始算起,再翻译16个终止(包含第1 024位),且这16个氨基酸的序列与原来的完全不同,对所编码蛋白质的功能影响比较明显。然而,我们未能获得胎儿的病理组织切片,从病理上进一步确认胎儿符合肾小管发育不良的诊断。
1983年Allanson等[14]报道了一孕妇2次因妊娠中期羊水过少而胎死宫内。2个胎儿各器官大体结构均未发现异常,而肾组织学病理特点为肾小管缺如、塌陷。此后,这种改变被命名为RTD。RTD患胎或患儿多于围生期死亡,重度羊水过少是该病的征兆,是由于胎儿尿量减少导致的。持续性羊水过少导致胎儿躯体压缩和活动受限,导致Potter序列综合征,表现为典型的面部异常、多余皮肤、肢体定位缺陷与支气管肺发育不良。对RTD患者进行超声检查,可见肾正常或略有放大,有或无肾实质回声异常或皮髓质分离。产后初期超声与孕期检查结果相似,且多未见胎儿生长受限[15-16]。部分早产新生儿无尿,伴顽固性低血压,多在出生后不久死于严重肾功能衰竭、低血压及支气管肺发育不良[17],另外部分RTD新生儿还有颅骨骨化缺陷,如大囟门和宽颅缝等表现[18]。
2005年,Gribouval等[18]研究了来自9个家庭的16例RTD患者。这些患者均在孕16~31周由超声发现羊水过少(其中2例是在妊娠28周以后发现羊水过少)。提取这些患儿及其父母的DNA进行基因检测,结果发现11例患者在涉及肾素-血管紧张素系统(renin-angiotensin system,RAS)的相关基因(1q32的REN基因、1q42的AGT基因、17q23的ACE基因和3q24的AGTR1基因)存在纯合或者复合杂合突变,而他们的父母是以上基因突变的携带者[18]。基于此,Gribouval等指出RTD是1种常染色体隐性遗传病,并推测胎儿无尿和肾损伤是由于RAS系统失活,导致肾长期处于低灌注状态,引起组织学改变。此外,RTD患儿的母亲孕期多有服用ACEI类药物的经历[19],这说明RTD与RAS可能存在着密不可分的联系。2012年,Gribouval等[20]再次报道了54例RTD患者的基因检测结果,他们来自48个无血缘关系的家庭。结果显示,这些患者均有明确的基因位点改变,其中64.4%的家庭ACE基因存在突变位点,20%的家庭存在REN基因突变,AGT和AGTR1基因突变占15.6%。近年来,有一些RTD胎儿或新生儿的基因检测结果报道,并报道了一些突变位点,如ACE基因c.798C>G和c.1486C>T突变[16,21];2015年Richer等[22]报道了ACE基因c.820_821delAG、c.3521delG等。但目前未检索到国内关于RTD的报道。
RTD是一种严重的肾结构紊乱,常导致胎死宫内或早期新生儿死亡。而单纯羊水过少可能是RTD在胎儿时期的唯一表现。行胎儿基因检测和引产后胎儿尸检,特别是肾组织病理学检查,有助于明确诊断。尤其是对于多次妊娠合并羊水过少的家庭,这些检查意义重大。
NPHP16家系临床表现和基因型分析符合遗传学共分离现象。突变c.621_c.622insTGGTG,在ANKS6基因的外显子2的第621位碱基与第622位碱基之间插入了序列为TGGTG的5个碱基,该突变为移码突变,引起的氨基酸改变:p.R208Wfs*5(NM_173551),该基因编码的氨基酸序列与原来完全不同,生物学危害性较高。而另一突变位点c.2394+1G>A(IVS13),为经典剪切位点突变,该突变会改变外显子的剪接方式,直接导致蛋白质的改变,生物危害性较高。
NPHP16一般于婴儿期或青少年期发病,发病后5 ~ 10年进展为终末期肾病,临床表现较多样,包括多囊性肾发育不良、肾功能不全、肾增大,可合并有肾外表现如主动脉瓣狭窄、肥厚型心肌病、动脉导管未闭、完全性内脏逆位、肺动脉狭窄、胆汁淤积、肝纤维化等。病变主要累及肾小管间质,肾小管萎缩,伴肾小管基底膜不规则变薄,间质纤维化,在皮髓交界处和髓质有多发囊肿形成,患者肾功能减退,最终发展为终末期肾病[23]。该病涉及的基因为ANKS6。2013年Hoff等[24]报道来自6个家庭的8例患者的病变,其中5个家庭中有婴儿期受累,进行性发展为终末期肾病,且有严重肾外的缺陷,包括梗阻性肥厚型心肌病、主动脉瓣狭窄、肺动脉狭窄、动脉导管未闭、内脏和门静脉周围的肝纤维化,而1个家庭则表现为青少年发病。在他们的研究中确定了6种不同ANKS6基因纯合突变、截断突变、剪接位点突变、错义突变。Hoff等[24]还发现在动物模型中,ANKS6基因突变会引起初级纤毛的改变导致发育异常,因此NPHP16还是一种初级纤毛疾病,其致病机制的研究还有待进一步揭示。本研究中的病例在胎儿期即有肾形态学的明显异常,同时伴有羊水过少,说明肾功能严重异常,同时也提示本研究中检测到的突变位点具有较高的致病性。
本次研究中采用的检查手段为尸检和病理,对于畸形的描述较精确,为CMA及WES数据分析提供准确的临床信息,使检测结果更加可靠。研究发现染色体微缺失重复与孕中期羊水过少未见相关性,CMA不是孕中期羊水过少必需的检查项目;妊娠中期发生羊水过少是某些遗传性疾病的表现,建议进行全外显子检测,帮助明确病因。
由于本研究中纳入的样本数量少,研究结果有待于进一步扩大样本量后得到进一步验证。此外本研究中发现的2例可疑致病位点均为国内外首次报道,接下来我们将对此进一步进行蛋白和功能验证。
综上,染色体微缺失重复与孕中期羊水过少未见相关性,CMA不是孕中期羊水过少必需的检查项目;部分妊娠中期羊水过少是由常染色体隐性遗传病引起,全外显子检测有助于明确病因,指导再次妊娠。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
中国生殖健康(2020年4期)2021-01-18 02:58:10
中国生殖健康(2018年4期)2018-11-06 07:12:16
西藏科技(2016年8期)2016-09-26 09:00:48
中国卫生标准管理(2015年7期)2016-01-15 03:58:37
中外医疗(2015年11期)2016-01-04 03:58:45
医学研究杂志(2015年8期)2015-06-22 14:00:57
医学研究杂志(2015年12期)2015-06-10 06:57:46
西南军医(2015年6期)2015-01-23 01:25:49
科学之友(2014年20期)2014-12-23 18:55:56
