
16-表雌三醇的合成及放射性标记
2018-07-23 08:47刘炳楠熊利平姜申德要少波
同位素 2018年5期
刘炳楠,熊利平,姜申德,要少波
(1.天津医科大学总医院 PET/CT影像诊断科,天津 300052;2.天津医科大学总医院 血液内科,天津 300052;3.天津大学 药物科学与技术学院,天津 300072;4.中国医学科学院 北京协和医学院 北京协和医院 核医学科,北京 100730)
1955年,Marrian成功从孕妇尿液中分离得到一种雌激素化合物,经结构表征后确定是16-表雌三醇[1],表雌三醇是在Marrian和Doisy从孕妇尿液中分离得的雌三醇后的又一种雌激素[2-3]。在随后的数十年中,又有许多种雌激素被发现并确定结构,例如16α-羟基雌酚酮[4]、16β-羟基雌酚酮和16-羰基-17β雌二醇[5-6]、18-羟基雌酚酮[7]等。16-表雌三醇具有甾体的化学结构,属于糖皮质激素,其主要临床应用是制备抗炎药物[8-10];并且,除了糖皮质激素作用以外,16-表雌三醇还是正电子断层显像(positron emission tomography, PET)研究中放射性探针18F-FES(18F-16α-fluoro-17β-estradiol)的合成前体[11],18F-FES在PET临床研究中主要用于特异性结合原发和继发乳腺癌肿瘤细胞中表达的雌激素受体水平,能够早期精准诊断乳腺癌[12-14]。因此在临床中,表雌三醇的用处很多。
迄今为止,16-表雌三醇主要通过化学合成,已知的表雌三醇合成方法主要有:在Huffman报道的合成路线中,以雌酚酮为起始原料经过5步反应,包括16-肟基衍生物制备、酯化反应、还原水解肟基和乙烯醇的氢化还原[15],这条合成路线较低的收率限制了表雌三醇的大规模生产;1957年,在Biggerstaff报道的合成路线中,主要包括四醋酸铅氧化和氢化锂铝还原反应[16],该路线用到了重金属铅化合物,对环境污染较大,还用到了易燃易爆的试剂氢化锂铝,该路线不适合大规模生产;1983年,Numazawa报道了另外一条合成路线,包括溴代反应、醇酮重排和PdCl2催化还原[17],该路线中用到的单质溴容易挥发,对实验条件要求较高,对环境尤其是空气污染较大,并且还用到了价格昂贵的钯试剂。
本研究报道了两条以雌酚酮为起始原料合成16-表雌三醇的简便合成路线,主要包括乙酰化、选择性保护、脱保护、氧化还原和重排等反应,反应条件温和、收率较高、所用试剂成本低,适合放大生产。同时,在对16-表雌三醇进行结构修饰后,完成了18F-FES的放射性标记和注射液的质量控制,经检测,制备的18F-FES注射液满足临床要求。
1 仪器与试剂
1.1 主要仪器
电磁搅拌器:德国IKA;熔点测定仪:奥地利Reichert-Thermovar;1H NMR谱数值测定仪Viarian INOVA(400 MHz或500 MHz):德国Bruker公司,使用四甲基(δ=0.00)作为内标,偶合常数用Hz作为单位,峰形分别记为s(singlet)、d(doublet)、t(triplet)、br(broad)和m(multiplet);快速硅胶柱色谱:300~400目柱层析硅胶;薄层层析色谱检测铝板(Kieselgel 60 F254):德国Merck公司,通过UV和高锰酸钾氧化显色;Cyclone RDS111加速器:德国Siemens公司;PET-MF-2V-IT-I型氟-18多功能合成模块:北京派特科技有限公司;Waters HPLC工作系统:美国Waters公司。
1.2 主要试剂
18-冠-6(18-Crown-6)、无水乙腈(MeCN,99.9%)、叔丁基二甲基氯硅烷(TBDMSCl):美国Sigma-Aldrich公司;乙酸乙酯、石油醚(60-90)、乙醇、二氯甲烷、醋酐、吡啶、醋酸、四氢呋喃、硼氢化钾、氢氧化钠、高氯酸:天津江天化工;标准品19F-FES:德国ABX公司;Sep-Pak light QMA柱和Sep-Pak plus C18柱:美国Waters公司。QMA柱的预处理(活化)依次用NaHCO3(8.4%水溶液)和注射用水冲洗;C18柱的预处理(活化)依次用乙醇和注射用水冲洗。
2 实验方法
2.1 16-表雌三醇的合成路线1
2.1.1烯醇二醋酸酯2的合成
对雌酚酮的双乙酰化步骤参照文献[18],合成路线示于图1,将雌酚酮(13.50 g,50 mmol)溶解于醋酸异丙烯酯(120 mL,1.1 mol),并加入对甲苯磺酸(4.05 g,23.5 mmol),搅拌下加热回流3.5 h。将反应溶液冷却,减压旋干溶剂,向反应瓶中加入乙酸乙酯300 mL将残余物溶解,依次用冷的5%碳酸氢钠和水洗,并用无水硫酸钠干燥,将有机溶剂旋干后即得到类白色固体,用乙酸乙酯重结晶得到白色固体12.5 g,收率73%,熔点147~149 ℃,文献149~150 ℃[18]。1H NMR(400 MHz,CDCl3)δ,0.94(s,3H,18-CH3),2.20(s,3H,-OCOCH3),2.30(s,3H,-OCOCH3),5.54(d,1H,J=1.3 Hz,16α-H),6.82-7.28(m,3H,aromatic)。鉴定为目标产物。
2.1.2环氧化物3的合成
将化合物2 (6.40 g,18 mmol)、70%间氯过氧苯甲酸(8.90 g,36 mmol)溶解在170 mL二氯甲烷中,置于室温搅拌2 h,反应完毕使用5%碳酸氢钠和水洗,并用无水硫酸钠干燥并将溶液蒸干后得到类白色固体,在乙酸乙酯中重结晶得到白色固体4.35 g,收率65%,熔点148~151 ℃,文献150~152 ℃[18]。1H NMR(400 MHz,CDCl3)δ,0.95(s,3H,18-CH3),2.13(s,3H,-OCOCH3),2.30(s,3H,-OCOCH3),3.94(s,1H,16β-H),6.81-7.28(m,3H,aromatic)。鉴定为目标产物。
2.1.3雌三醇4的合成
向含有5.83 g环氧化物3的乙醇溶液116 mL,并缓慢加入硼氢化钾(1.20 g,22.2 mmol)和氢氧化钾(5.03 g,89 mmol)的24 mL水溶液,将混合溶液置于室温搅拌反应1 h,减压旋蒸除去大部分乙醇,并加入50 mL 5%稀盐酸溶液,并用80 mL乙酸乙酯萃取3次,将乙酸乙酯溶液合并后用无水硫酸钠干燥并旋干得到固体,并用乙酸乙酯重结晶得到白色固体即为雌三醇3.63 g,收率80%,熔点275~280 ℃,文献279~284 ℃[19]。1H NMR(400 MHz,MeSO-d6)δ,0.67(s,3H,18-CH3),3.79-3.88(m,1H,16α-OH),4.59(d,1H,J=5.1 Hz,17α-H),4.66(d,1H,J=4.8 Hz,16β-H),6.43-7.04(m,3H,aromatic),7.96(s,1H,17β-H),8.97(s,1H,3-OH)。鉴定为目标产物。
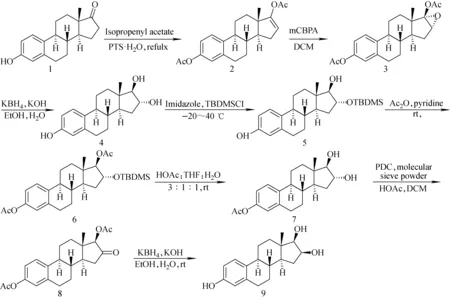
图1 16-表雌三醇的合成路线1Fig.1 The first synthetic route of 16-epiestriol
2.1.416α-叔丁基二甲基硅基雌三醇5的合成
选择性保护16α-羟基的实验步骤参考已发表文献方法[19-20],将雌三醇(2.00 g,6.9 mmol)和咪唑(1.10 g,16.6 mmol)溶于36 mL DMF中,冷却至-40 ℃搅拌下逐滴加入溶有叔丁基二甲基氯硅烷(2.30 g,15.3 mmol)的10 mL DMF溶液,加毕在此温度下搅拌反应1.5 h,随后将反应溶液倒入冰水中静置过夜,抽滤并收集沉淀,将沉淀复溶于50 mL乙酸乙酯中,乙酸乙酯溶液用水洗并用无水硫酸钠干燥,减压旋干溶剂后得到灰白色固体,用快速硅胶柱色谱分离得到白色固体2.2 g,收率79%,熔点192~195 ℃,文献194 ℃[20]。1H NMR(400 MHz,CDCl3)δ,0.11(d,6H,J=2.1 Hz,-Si(CH3)2),0.81(s,3H,18-CH3),0.94(s,9H, -SiC(CH3)3),2.83(d,1H,J=3.8 Hz,17β-OH),3.59(d,1H,J=4.8 Hz,17α-H),4.09-4.13(m,1H,16β-H),4.59(s,1H,3-OH),6.58-7.17(m,3H,aromatic)。鉴定为目标产物。
2.1.53,17β-二乙酰-16α-叔丁基二甲基硅基雌三醇6
将化合物5(4.09 g,10.2 mmol)溶于16.1 mL吡啶和8 mL醋酐中,置于室温搅拌过夜,将溶液倒入冰水中待沉淀充分析出过滤,将沉淀溶于100 mL乙酸乙酯中依次用水洗和无水硫酸钠干燥,减压旋干溶剂后使用快速硅胶柱分离得到白色固体4.7 g,收率95%,熔点108~110 ℃,文献110~112 ℃[19]。1H NMR(400 MHz,CDCl3)δ,0.05(d,6H,J=5.6 Hz, -Si(CH3)2),0.80(s,3H,18-CH3),0.90(s,9H,-SiC(CH3)3),2.11(s,3H,-OCOCH3),2.30(s,3H,-OCOCH3),4.30-4.33(m,1H,16β-H),4.86(d,1H,J=5.6 Hz,17α-H),6.81-7.28(m,3H,aromatic)。鉴定为目标产物。
2.1.63,17β-二乙酰基-16α-雌三醇7
选择性脱除硅保护基过程参照文献报道[19,21],将化合物6溶解于141 mL醋酸-四氢呋喃-水(3∶1∶1)溶液中,室温搅拌24 h,加入200 mL乙酸乙酯并用碳酸氢钠洗和无水硫酸钠干燥,减压蒸干后得到类白色固体,通过快速硅胶柱色谱分离得到白色固体1.58 g,收率92%,熔点136~138 ℃,文献132 ℃[20]和140 ℃[19]。1H NMR(400 MHz,CDCl3)δ,0.88(s,3H,18-CH3),2.17(s,3H,-OCOCH3),2.30(s,3H,-OCOCH3),3.73(s,1H,16α-OH),4.14-4.19(m,1H,16β-H),4.30(d,1H,J=4.1 Hz,17α-H),6.81-7.29(m,3H,aromatic)。鉴定为目标产物。
2.1.73,17β-二乙酰基-16-酮化合物8
氧化反应依照经典条件进行[22],将化合物7(1.47 g,3.9 mmol)、1.8 g分子筛粉末、PDC(1.9 g,4.9 mmol)[23]加入18 mL二氯甲烷中,逐滴加入醋酸0.3 mL,将反应物置于室温搅拌1.5 h,加入水稀释并用60 mL乙酸乙酯萃取2次,合并有机相用无水硫酸钠干燥后减压蒸干溶剂得到棕色沉淀,使用快速硅胶柱色谱纯化得到白色固体1.15 g,收率79%,熔点130~132 ℃,文献133.5~135.5 ℃[24]。1H NMR(400 MHz CDCl3)δ,0.88(s,3H,18-CH3),2.21(s,3H,-OCOCH3),2.30(s,3H,-OCOCH3),5.13-5.15(m,1H,17α-H),6.84-7.31(m,3H,aromatic)。鉴定为目标产物。
2.1.816-表雌三醇9
还原反应过程类似于制备雌三醇4的过程,还原反应完毕后使用甲醇重结晶得到白色固体粉末,收率81%,熔点285~287 ℃,文献281~289 ℃[16]。1H NMR(500 MHz,Me2SO-d6))δ,0.72(s,3H),4.05-4.28(m,1H,17α-H),4.55(br,1H,16α-H),6.42-7.02(m,3H,aromatic),9.00(s,1H,3-OH)。鉴定为目标产物。以雌酚酮为起始原料计,总收率19%。
2.2 16-表雌三醇的合成路线2
2.2.1化合物10的合成
合成步骤示于图2,环氧化物的开环反应依照文献中报道步骤[18],将化合物3(1.00 g,2.7 mmol)、6.4 mL醋酸和混合酸(醋酸:高氯酸4∶1)混合后置于室温搅拌反应2 h,加入100 mL乙酸乙酯并用冷的5%碳酸氢钠和水洗,无水硫酸钠干燥后旋干后得到棕红色油状物,加入醋酐和吡啶完成乙酰化反应,结束后萃取并旋干溶剂,用快速硅胶柱色谱分离,得到化合物10 0.89 g,收率89%,熔点164~167 ℃,文献168~171 ℃[16]。1H NMR(400 MHz,CDCl3)δ,1.01(s,3H,18-CH3),2.14(s,3H,-OCOCH3),2.29(s,3H,-OCOCH3),5.46(d,1H,J=8.8 Hz,16β-H),6.82-7.29(m,3H,aromatic)。鉴定为目标产物。
2.2.2化合物8的合成
甾体D环的开环反应依照参考文献中方法[18,25-26],将化合物10(0.89 g,2.4 mmol)溶于26.8 mL甲醇和13.2 mL 5%氢氧化钾水溶液中,室温下搅拌反应5 h,将反应溶液倒入50 mL 2%稀盐酸溶液中,并用80 mL乙酸乙酯萃取2次,合并有机相用无水硫酸钠干燥并旋干,经醋酐和吡啶乙酰化后用快速柱色谱分离,得到白色固体0.54 g,收率61%。
2.2.3由化合物8制备表雌三醇
该合成步骤同2.1.8中描述,最终得到表雌三醇。以雌酚酮为起始原料计,总收率22%。
2.3 18F-FES前体合成与标记
2.3.1化合物11的合成
合成步骤示于图3,将化合物9(50 mg,0.14 mmol),1 mol/L氢氧化钾水溶液(0.14 mL,0.14 mmol),10 mL乙腈混合后搅拌反应10 min,得到白色的浑浊反应体系,旋蒸蒸干溶剂,再加入乙腈10 mL,蒸干,重复3次,向单口烧瓶中加10 mL乙腈,滴加甲氧基氯甲烷(20 mg,0.16 mmol),常温搅拌12 h,减压旋干溶剂,加水20 mL,15 mL乙酸乙酯萃取3次,合并有机相并用无水硫酸钠干燥,经快速柱色谱纯化得到白色的固体中间体。将中间体和硫酰二咪唑溶解于干燥四氢呋喃中,并在冰浴条件下缓慢加入氢化钠,室温反应2 h后处理并经快速柱色谱分离纯化,得到白色固体11。1H NMR(CDCl3):7.18(d,J=8.5 Hz,1H,H-1),6.85(dd,J=8.5 Hz,2.5 Hz,1H,H-2),6.79(d,J=2.5 Hz,1H,H-4),5.16(m,overlapping,1H,H-16),5.15(s,2H,OCH2),4.60(d,J=7.5 Hz,1H,H-17),3.48(s,3H,OCH3),2.95-2.84(m,2H),2.50-2.24(m,overlapping,3H),2.14-2.10(m,1H),1.92-1.80(m,2H),1.68-1.54(m,3H),1.02(s,3H,18-CH3)。鉴定为目标产物。
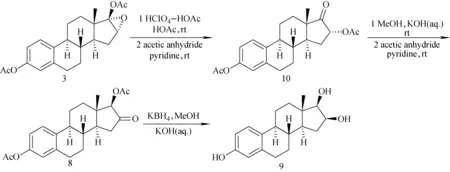
图2 16-表雌三醇的合成路线2Fig.2 The second synthetic route of 16-epiestriol
2.3.218F-FES的放射性标记
回旋加速器通过18O(p,n)18F核反应生产得到18F离子,经过QMA柱俘获后用18-Crown-6/KHCO3的乙腈-水混合溶液1 mL淋洗到反应瓶中;通氮气(100 mL/min)并在116 ℃加热条件下将液体蒸发至干燥;加入1.5 mL无水乙腈,通氮气并加热至116 ℃,使残留的水和乙腈共沸再次除水;将5 mg前体11的乙腈溶液加入至含有活性[18F]KF/18-Crown-6/KHCO3的反应管,在封闭的反应管中加热至100 ℃并保持20 min;鼓氮气将反应管中的乙腈在80 ℃下蒸干,将0.1 mol/L稀盐酸溶液加入至反应管,加热至140 ℃保持15 min,再次加热至80 ℃下鼓氮气除去混合溶液中的乙醇;使用半制备液相色谱进行分离纯化,流速为5 mL/min,流动相为50%(乙醇/水),收集产物峰(保留时间为7 min左右)并加入适量生理盐水稀释使乙醇含量小于10%,将含有18F-FES的注射液的通过0.22 μm的无菌滤膜至无菌产品瓶中,18F-FES注射液使用radio-HPLC分析其放化纯度,Waters系统分析型HPLC: Waters C18柱,4.6 mm×250 mm;流速:1 mL/min;流动相:乙醇∶水=1∶1。
3 结果与讨论
3.1 化学合成
两条表雌三醇的合成路线中,以雌酚酮为起始原料,经过简单的反应,分别得到19%和22%表雌三醇,各步反应产物经过了熔点和1H NMR谱确定其结构身份。反应简单、收率较高,较适合放大生产。
3.2 放射性标记
以化合物11为前体,经过两步简单的氟化和水解反应,产物并经过半制备HPLC分离纯化即得。18F-FES注射液可在60 min内完成制备,注射液为无色澄清透明的溶液,pH为6.5~7.5,放化收率(30±4)%(n=7),比活度(1.75±0.25) Ci/μmol。注射液经过分析型radio-HPLC检测,保留时间为11.9 min,放化纯度>99%(图4),满足临床要求,与标准品19F-FES经HPLC检测的紫外谱图对比保留时间一致,可确定合成产物为18F-FES。
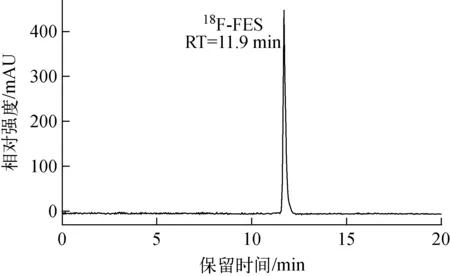
图4 18F-FES注射液的radio-HPLC分析Fig.4 Radio-HPLC analysis of 18F-FES injection
3.3 合成中无法进行的化学反应步骤
对甾体环的16α-羟基进行结构修饰和反应时,总结了几种不成功的反应,具体示于图5。
3.3.116α-羟基硅保护基的脱保护
在脱除化合物6的16α-羟基的叔丁基二甲基硅基保护基时,最初尝试经典方法四丁基氟化铵-四氢呋喃系统[20-21,27-29],发现并不奏效,即使时间延长至48 h,仍然没有发生反应(通过TLC检测),与Laurent等[20]报道的实验结果相悖,因此尝试了Wu等[19]报道的方法,使用醋酸四氢呋喃-水的体系,该反应在温和的条件下完成。
3.3.216α-羟基的Mitsunobu反应
当成功得到化合物7时,曾想是否可以通过Mitsunobu反应[30-31]直接将16α-羟基构型转化变为16β-羟基,可直接得到表雌三醇。然而在尝试反应过程中,亲核试剂分别试过对甲苯磺酸和苯甲酸,搅拌10 h反应仍未发生。
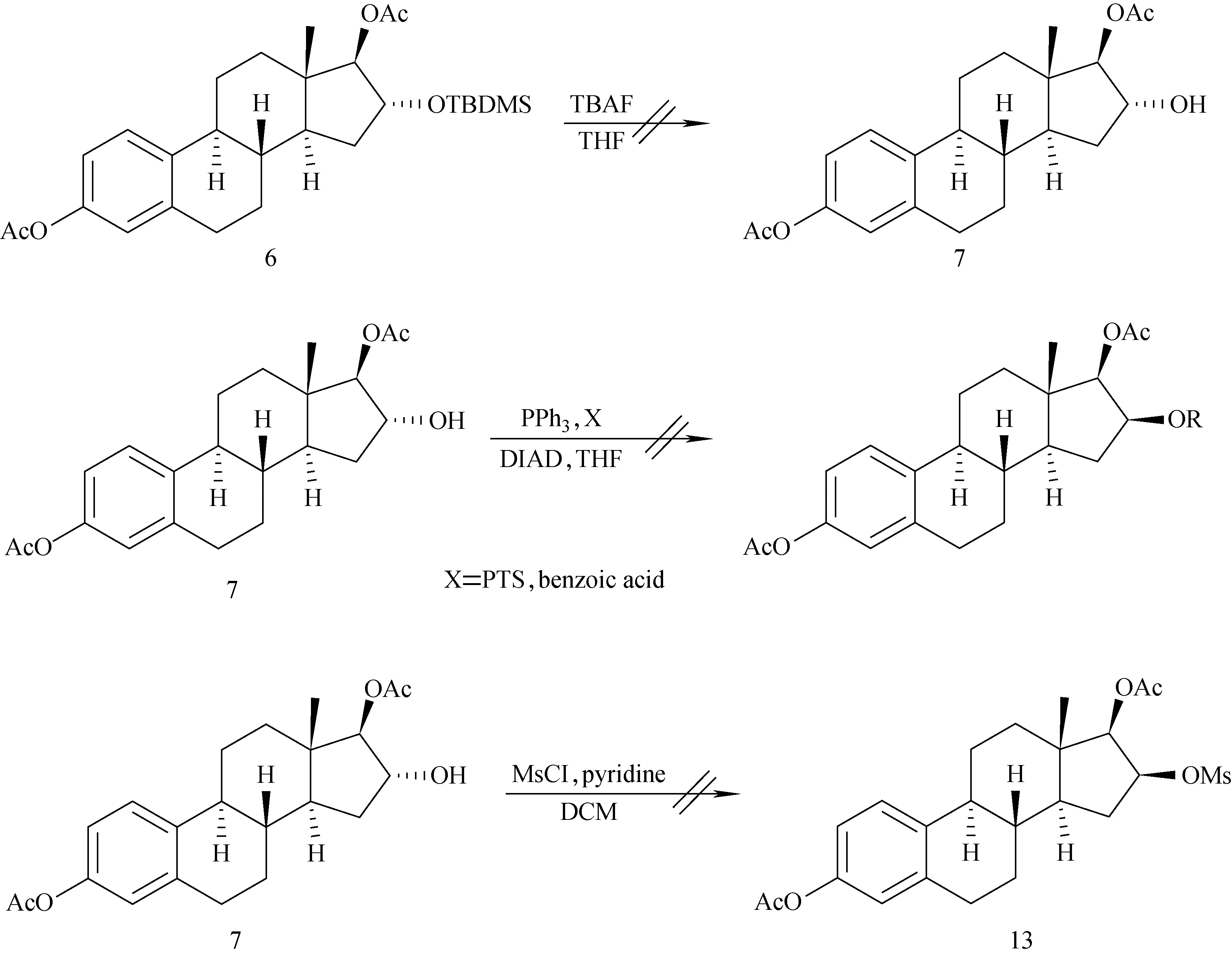
图5 实验中无法进行的反应Fig.5 Representative failed reactions
3.3.316α-羟基的甲磺酰化
对化合物7的16α-羟基尝试甲磺酰化反应,可以通过亲核氟化反应,直接完成标记,进而得到16β-18F-FES,可以探究16β-18F-FES和18F-FES两种雌激素结构类似物在体内的生物学活性差异,然而该磺酰化反应并未进行,反应过夜仍未发现产物。
总结以上三种针对16α-羟基进行的反应可以看出,由于18位甲基的空间位阻较大,导致针对16α-羟基进行的反应不容易发生,也就是不够活泼,因此不能完成理想反应。
4 结论
本文中以雌酚酮为原料,成功完成两种表雌三醇的合成路线,收率较高、产率稳定,条件温和,适合放大生产。并以表雌三醇为前体进行结构修饰,成功制备满足临床要求的雌激素受体类PET显像剂18F-FES,可为临床使用提供有力支持。
猜你喜欢
盐科学与化工(2022年3期)2022-04-11
昆明医科大学学报(2021年8期)2021-08-13
环境科学研究(2020年3期)2020-03-17
中成药(2017年8期)2017-11-22
中成药(2017年5期)2017-06-13
中成药(2017年3期)2017-05-17
浙江化工(2017年4期)2017-05-11
人间(2015年11期)2016-01-09
浙江畜牧兽医(2014年5期)2014-02-27
数理化学习·高一二版(2009年1期)2009-03-19
