
欧洲儿童肝豆状核变性诊疗推荐意见简介
2018-07-17 03:51曹海霞范建高
实用肝脏病杂志 2018年4期
曹海霞,范建高
作者单位:200092上海市 交通大学医学院附属新华医院消化内科
肝豆状核变性(Wilson’s disease,WD)是常染色隐性遗传相关的铜代谢障碍性疾病,估计患病率为1:30000。该病是由于编码P型ATP酶的ATP7B基因突变,影响铜的分泌及排泄所致铜沉积。儿童肝豆状核变性的临床表现可以从无症状肝脏疾病到肝硬化或者肝衰竭,但是神经精神症状却非常少见。常规的检测方法包括血清铜蓝蛋白、24 h尿铜。最终的诊断需要基于症状、评估铜代谢的生化学检查和ATP7B基因突变的诊断评分系统。该病的治疗需要药物终身治疗,可以使用螯合剂青酶胺或者曲恩汀减少体内过多的铜或者锌剂减少肠道铜的吸收。急性肝衰竭患者常常需要肝移植。儿童时期WD的诊断非常困难,经常是无症状的,成人推荐的标准可能不适合。因此,欧洲儿童胃肠病、肝病及营养学会(ESPGHAN)推出这篇文章,目的是为儿童肝豆状核变性的诊断、治疗及随访提供推荐意见。
本文通过搜索MEDLINE,EMBASE,Cochrane数据库中从1990年到2016年关于儿童WD的所有前瞻性和回顾性的研究,根据GRADE系统评估证据的可靠性。如果证据弱,则将专家的意见作为推荐意见。最终,ESPGHAN核心小组和ESPGHAN肝病学会成员对每个推荐意见进行无记名投票。关于儿童肝豆状核变性的诊断流程、推荐意见、常见表现和评分系统简介如下:
1 肝豆状核变性的诊断流程
见表1。

表1 肝豆状核变性诊断流程
2 ESPGHAN肝病学会的推荐意见
2.1 对1岁以上患儿出现无症状的转氨酶升高、肝硬化合并肝脾肿大或者腹水、急性肝衰竭等以肝病为表现者都应排除WD。
2.2 青少年患者出现不能解释的认知障碍、精神障碍或者运动障碍需排除WD。
2.3 疑诊WD的患者应该完善肝功能检查【包括ALT、AST、TBIL、DBIL、ALP及凝血酶原时间(PT)/INR】及铜蓝蛋白和24 h尿铜。
2.4 Ferenci评分系统可以用于儿童WD的诊断。ATP7B基因突变分析有助于WD的诊断。
2.5 不能确诊的儿童WD行肝铜检测有助于诊断。
2.6 一旦确诊WD,患者的一级亲属包括兄弟姐妹、后代及父母均应行肝功能、铜代谢指标及基因检测。
2.7 建议锌剂治疗用于家庭筛查发现的症状前的儿童WD或者螯合剂长期治疗后转氨酶正常的WD患者的维持治疗,从药物安全性角度考虑,首选醋酸锌。
2.8有明显肝病表现如肝硬化及异常INR的患者,建议使用螯合剂治疗。
2.9 使用螯合剂治疗的患者,建议控制食物中铜的摄入,直至患者症状减轻、转氨酶正常。
2.10 建议将急性肝衰竭及失代偿期肝硬化儿童WD转诊到儿童肝移植中心进一步诊治。
2.11 对于失代偿期肝硬化儿童,建议使用螯合剂或者螯合剂联合锌剂治疗,对药物治疗应答者可以不行肝移植治疗。应用King’s Wilson指数可以用于评估肝移植的时机及预后。
2.12 肝移植可以纠正铜代谢酶的异常,因此肝移植后不再需要螯合剂或者锌剂治疗。
2.13 所有儿童在开始治疗的第1月内均应密切随访,以后每1~3月随访直至疾病缓解,随后可以每3~6月随访。
2.14 随访项目包含:体格检查、生化学检查(血常规、肝功能、肾功能、尿蛋白)、血清铜、24 h尿铜,主要用于评估治疗的效果、药物剂量是否足够、患者的依从性及药物的副作用。
2.15 患者服用锌剂的依从性可以通过检测血锌水平、尿锌、24 h尿铜来评估。
2.16 如果患者转氨酶仍较高或者治疗后有反弹,提示患者在药物治疗方面的依从性不好。
2.17 一旦发生青霉胺相关的副作用,建议立即停药。根据肝病的严重程度调整为曲恩汀或者锌剂治疗。
3 儿童肝豆状核变性患者的临床表现及评分系统等
见表2~表6。

表2 儿童肝豆状核变性患者的临床表现

临床表现 发病年龄眼科表现*裂隙灯下可见K-F环 >10岁血液系统* 急性/慢性溶血 >7岁其他系统的表现肾脏* 肾小管功能障碍(Fanconi’s综合征、肾小管酸中毒、氨基酸尿)*肾结石*肾钙质沉着心脏*心肌病,心功能不全*心律失常内分泌*甲状旁腺功能减退其他*胰腺炎*皮肤脂肪瘤骨骼*骨质疏松/佝偻病*关节病发病年龄不确定,仅限于病例报道
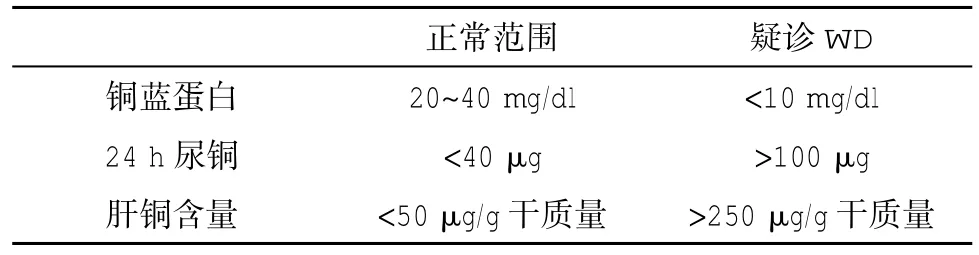
表3 铜代谢相关指标

表4 肝豆状核变性诊断评分系统

表5 常用药物的治疗剂量及随访监测指标
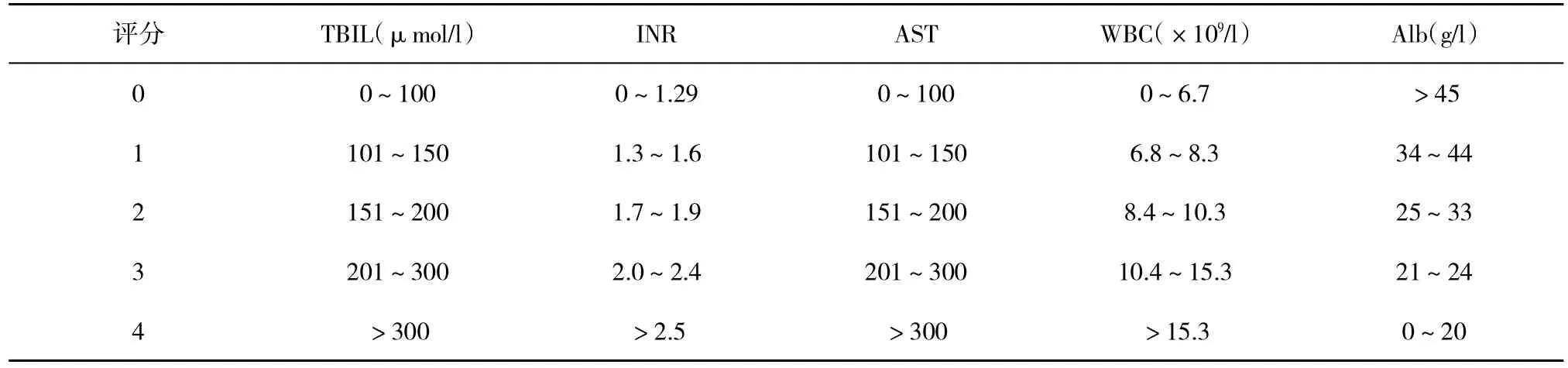
表6 儿童肝豆状核变性致失代偿期肝病的预后评分系统(King’s Wilson index)
猜你喜欢
环境科学研究(2022年10期)2022-10-19
中南民族大学学报(自然科学版)(2022年3期)2022-05-08
中国土壤与肥料(2021年5期)2021-12-02
现代临床医学(2021年4期)2021-07-31
华人时刊(2020年21期)2021-01-14
中兽医学杂志(2019年1期)2019-01-06
罕少疾病杂志(2016年4期)2016-03-11
医学综述(2015年17期)2015-02-10
中国药业(2014年19期)2014-05-17
中华移植杂志(电子版)(2012年3期)2012-08-15
