
壳聚糖包覆姜黄素脂质体体外释放和药代动力学研究
2018-06-08 09:24余忠姝张景勍
中国药理学通报 2018年6期
李 嫄,赵 静,余忠姝,曾 梅,张景勍
(重庆医科大学药学院重庆高校药物工程研究中心,重庆 400016)
多年生草本姜黄素(curcumine, CM)是一种从姜黄根茎中提取的酚类活性物质,广泛种植于热带南亚和东南亚国家[1]。许多研究表明其毒性低,在抗肿瘤方面的潜力巨大[2]。CM对所有类型的肺癌细胞具有细胞毒性作用,如非小细胞肺癌A549和H1975细胞、大细胞肺癌H460细胞、肺鳞状细胞癌Calu-1细胞、小细胞肺癌H446和H187[3]。然而,水溶性差和快速降解引起生物利用度低,因此,提高CM的生物利用度对临床应用具有重要意义[4]。脂质体是由磷脂双分子层形成的、内部包含药物的封闭囊泡,具有缓释、靶向性、组织相容等优点。壳聚糖为天然高分子多糖,具有良好的生物相容性、可降解性和黏附性[5-6],可用于脂质体的包衣,以增加脂质体的稳定性和靶向性。近年来,壳聚糖在医药领域中的应用不仅包括止血、抗菌、人造组织器官等研究,还被广泛用作药物载体材料[7-8]。本文初步考察了壳聚糖包衣姜黄素脂质体(chitosan coated curcumin liposomes, CMLP-CS)的体外释药特征及大鼠体内的药代动力学行为,以期促进CM吸收,提高其生物利用度,并为进一步研究CMLP-CS口服给药奠定基础。
1 材料
1.1药物与试剂CM(规格:10 mg,西安帅诺生物科技有限公司);蛋黄卵磷脂(规格:500 mg,国药集团化学试剂有限公司);胆固醇(规格:500 mg,国药集团化学试剂有限公司);尼群地平(纯度>99%,国药集团化学试剂有限公司);壳聚糖(脱乙酰度95%,规格1 g,浙江金壳生物化学有限公司);乙腈、乙醇等均为色谱纯,实验用水均为超纯水。
1.2仪器LC-2010AHT高效液相色谱仪(日本岛津公司);T-6新世纪紫外分光光度计(北京普析通用仪器有限责任公司);Nano-ZS90型马尔文粒径测定仪(英国马尔文公司)。
1.3实验动物12只SD健康大鼠,♂,体质量(230±20)g,动物使用许可证号:SYXK(渝)2015-0001,重庆医科大学动物实验中心提供。
2 方法
2.1CMLP-CS的制备采用薄膜分散法制备姜黄素脂质体[9]。称取处方量的磷脂、胆固醇和姜黄素于梨形瓶中,用无水乙醇超声溶解后,旋转蒸发挥去乙醇,在瓶壁形成均匀类脂薄膜。再加入适量磷酸盐缓冲液 (pH 6.8 PBS),45℃旋转水合60 min使膜溶解,探头超声5 min,即得姜黄素脂质体。将得到的上述脂质体初悬液10 mL,缓慢滴加到一定体积的2 g·L-1CS溶液中,再继续搅拌120 min(700 r·min-1),冷却至室温,4℃冰箱静置保存。
2.2体外释放动力学动态透析法考察CMLP-CS和CM的体外释药特点。将同浓度的CMLP-CS和CM各1 mL分别投进预先处理好的透析袋,将透析袋分别投入两种释放介质(pH 1.2 HCl; pH 6.8 PBS)中,平行操作3份,于水浴恒温震荡(37℃,100 r·min-1),分别于不同时间点取出1 mL释放介质,同时补充等温等体积的释放介质[10]。测定取出的释放介质中CM的紫外吸光度,计算CM浓度,绘制释放曲线,并采用相似因子法对释放曲线进行评价。
2.3大鼠体内药代动力学
2.3.1给药方案与样品采集 将♂ SD大鼠禁食12 h,自由饮水,随机分为2组,分别灌胃给予CMLP-CS、CM(给药剂量均为45.0 mg·kg-1),给药后于不同时间点大鼠眼眶取血500 μL,6 000 r·min-1离心10 min(离心管已经过肝素预处理),取上层血浆于-20℃冰箱保存。
2.3.2血浆样品处理方法 取200 μL待测血浆样品,加入80 μL尼群地平内标工作液(5 mg·L-1),乙酸乙酯萃取2次,每次加入500 μL乙酸乙酯,涡旋震荡5 min后,12 000 r·min-1离心10 min,合并有机相后用氮气挥干,加入100 μL流动相复溶,吸取20 μL复溶液进样[12]。
2.3.3样品测定方法的建立 色谱柱为伊利特C18色谱柱(250 mm×4.6 mm, 5.0 μm);流动相为乙腈 ∶5%冰乙酸溶液=55 ∶45(V/V);流速为1.0 mL·min-1;检测波长为430 nm;柱温为30℃。称取CM 10 mg,乙醇溶解并定容至50 mL,得200 mg·L-1CM储备液。取空白血浆200 μL,用CM储备液配制浓度分别为15、25、50、75、100、200、400 μg·L-1的CM系列血浆溶液,各取20 μL进样记录峰面积[12]。以CM和内标的峰面积之比为纵坐标Ss/Si,以CM浓度为横坐标c,做线性回归,并计算其回归方程。
2.3.4方法学考察 取空白血浆,制备高、中、低(400、200、50 μg·L-1)3个浓度的CM血浆样品,考察精密度,萃取回收率和方法回收率[12]。
2.3.5数据分析 通过DAS 2.1.1软件计算和分析数据,对CMLP-CS和CM的AUC(0~72h)及cmax等主要药动学参数进行方差分析,进行90%可信限考察,Tmax采用非参数统计Wilcoxon检验,评价CMLP-CS和游离CM是否具有生物等效性(α=0.05)。
3 结果
3.1体外测定方法经考察,制备的CMLP-CS的平均粒径约为550 nm,平均Zeta电位为23 mV,包封率约为78%。体外标准曲线方程为Y=0.1468X-0.0094,R=0.9999。CM溶液的浓度在0.5~8 mg·L-1范围内线性良好。日内精密度RSD分别为2.58%、2.03%、1.93%,日间精密度RSD分别为2.67%、3.44%、2.91%,均符合方法学要求。
3.2体外释放动力学
3.2.1体外释放曲线 CMLP-CS和CM在两种释放介质中的体外释放曲线,见Fig 1。CMLP-CS在pH 1.2 HCl和pH 6.8 PBS中,48 h内为快速释放期,累积释放率分别为(64.99±0.65)%和(63.34±1.23)%,48 h后缓慢并持续释放。在148 h时,CMLP-CS在pH 1.2 HCl和pH 6.8 PBS中的累积释放率分别为(70.48±0.50)%和(72.35±1.04)%,分别是游离CM的1.8倍和2.6倍。结果表明,CMLP-CS明显提高了体外释放率,可改善体外释药行为。
3.2.2溶出曲线评价 采用相似因子法[14]评价溶出曲线,经计算得到的相似因子见Tab 1。CMLP-CS与CM分别在pH 1.2 HCl或pH 6.8 PBS中的相似因子小于50,提示释放行为不相似,两者之间没有明显差异。此外,CM在pH 1.2 HCl或pH 6.8 PBS中的相似因子大于50,CMLP-CS在pH 1.2 HCl或pH 6.8 PBS中的相似因子大于50,提示它们在pH 1.2 HCl或pH 6.8 PBS中释放行为相似,而且两者之间没有差异,上述结果与Fig 1描述一致。
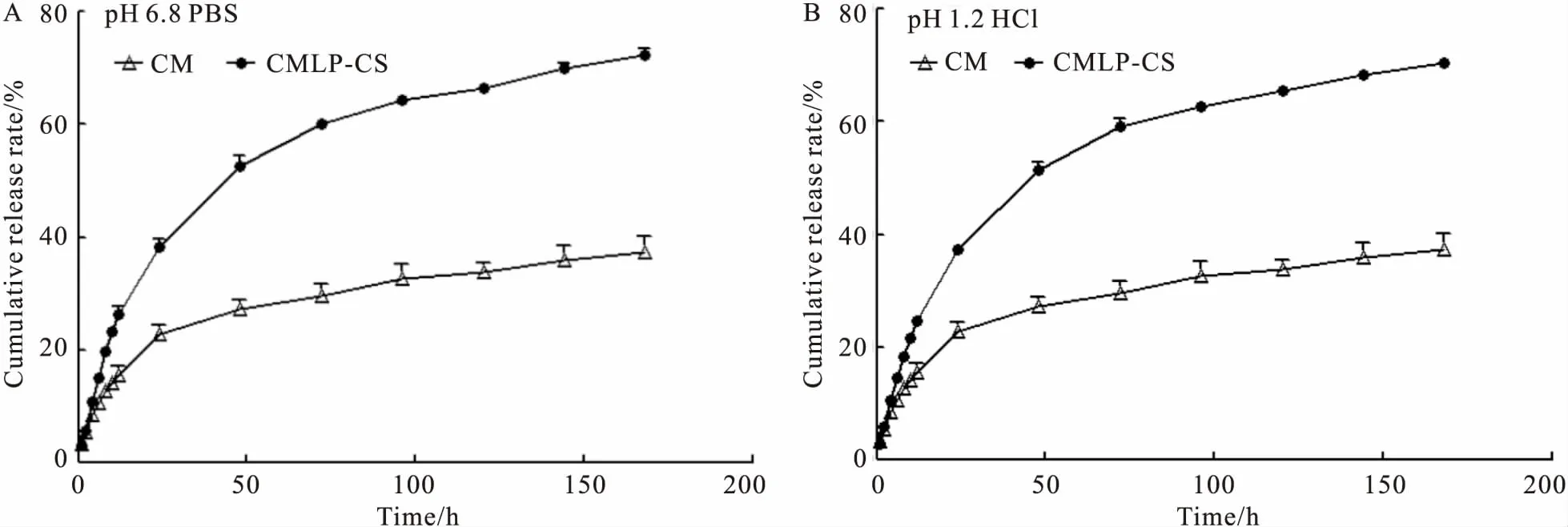
Fig 1 Drug release curve of CMLP-CS and CM in pH 6.8 PBS(A) and pH 1.2 HCl(B)
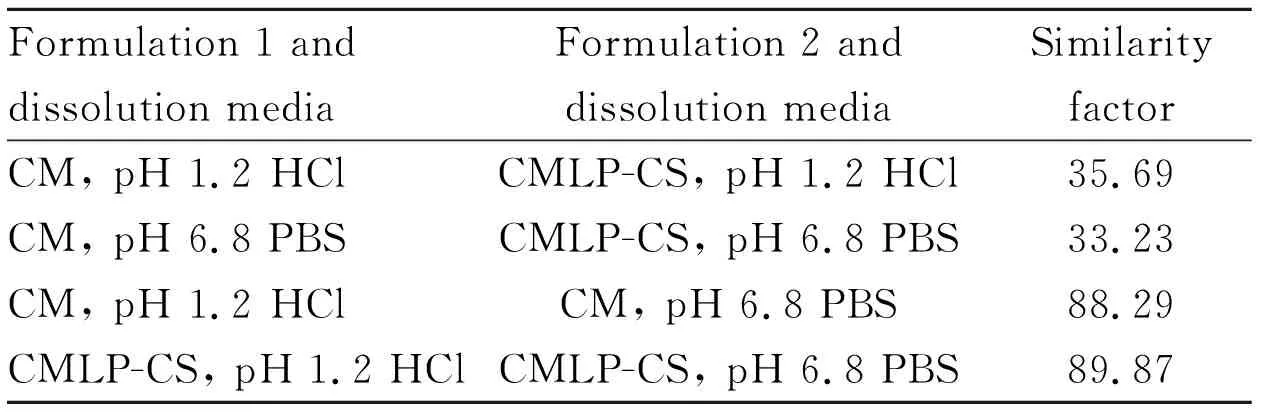
Formulation 1 anddissolution mediaFormulation 2 and dissolution mediaSimilarityfactorCM, pH 1.2 HClCMLP-CS, pH 1.2 HCl35.69CM, pH 6.8 PBSCMLP-CS, pH 6.8 PBS33.23CM, pH 1.2 HClCM, pH 6.8 PBS88.29CMLP-CS, pH 1.2 HClCMLP-CS, pH 6.8 PBS89.87
3.3大鼠体内药代动力学研究
3.3.1测定方法的建立 对浓度c与S(Ss/Si,待测血浆样品和内标物的峰面积之比)进行线性回归,得回归方程为:S=0.010 5c+0.123 3,r=0.9970,CM在15~400 μg·L-1浓度范围内线性良好,最低检测限(S/N=3)为5 μg·L-1。该方法在CM低、中、高3个浓度的日内精密度RSD和日间精密度RSD均小于5%,均符合生物样品分析要求[12],其萃取回收率和方法学回收率符合含量测定方法要求。
3.3.2药动学参数 CMLP-CS和游离CM的平均血药浓度-时间曲线,见Fig 2。经DAS 2.1.1软件计算分析,主要药动学参数见Tab 2。Fig 2显示,CM和CMLP-CS分别在0.25 h和3 h时达到了最大血药浓度,可知CMLP-CS的达峰时间Tmax是CM的12倍,CMLP-CS的cmax值也明显大于CM,说明与CM相比,CMLP-CS具有更长的释放时间,表现出缓释作用。CMLP-CS的曲线下面积AUC远远大于CM,表明CMLP-CS其在体内的吸收效果明显优于CM,口服生物利用度更好。由Tab 2计算可知,CMLP-CS的AUC(0~72h)值约为CM的8.9倍,CMLP-CS的相对生物利用度为846.5%,CMLP-CS明显提高了CM的体内口服生物利用度。CMET的MRT(0~72h)大于CM,其值为CM的3.7倍,说明CMLP-CS能够克服CM在体内被迅速消除的缺点,延长在体内的作用时间,从而利于提高生物利用度。
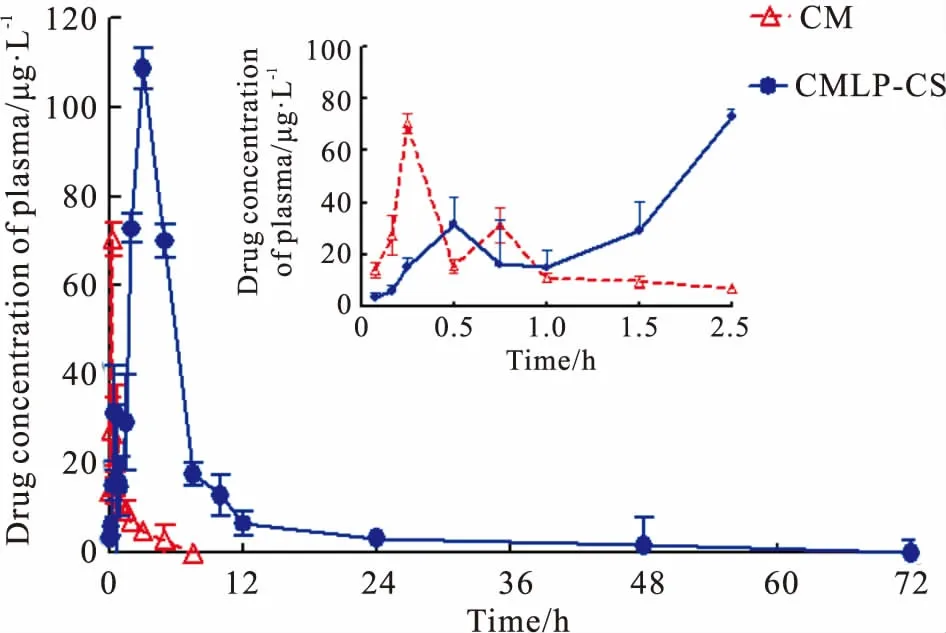
Fig 2 Mean concentration-time curves of CMLP-CS andfree CM with oral administration (n=6)
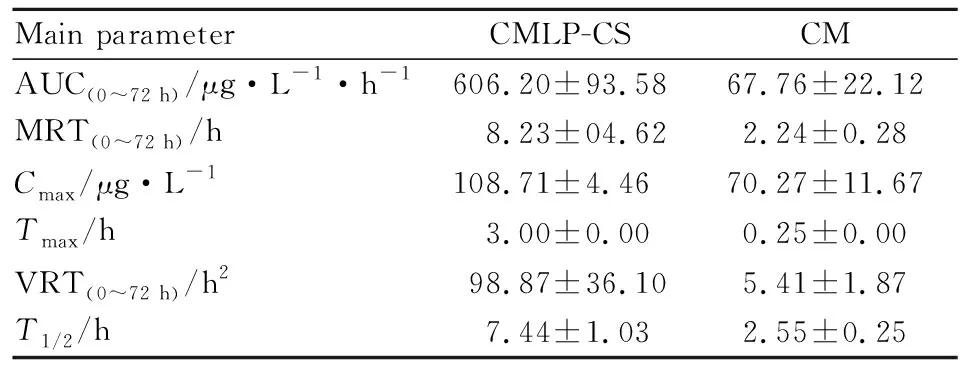
Main parameterCMLP-CSCMAUC(0~72 h)/μg·L-1·h-1606.20±93.5867.76±22.12MRT(0~72 h)/h8.23±04.622.24±0.28Cmax/μg·L-1108.71±4.4670.27±11.67Tmax/h3.00±0.000.25±0.00VRT(0~72 h)/h298.87±36.105.41±1.87T1/2/h7.44±1.032.55±0.25
3.3.3生物等效性 由Tab 3可知,CMLP-CS与游离CM的AUC(0~72 h)、AUC(0~∞)两个参数的90%可置信区间均不在生物等效性标准区间范围内,因此CMLP-CS与游离CM生物等效性不合格。另外,对达峰时间Tmax进行非参数统计Wilcoxon检验,结果显示,CMLP-CS和CM的达峰时间Tmax差异具有显著性(P<0.05)。按照生物等效性的判定标准,CMLP-CS与CM生物不等效,CMLP-CS的药效学标准高于CM。
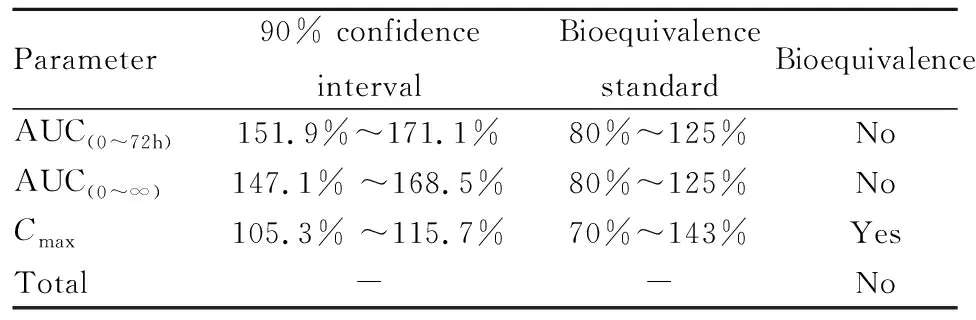
Tab 3 The bioequivalence comparison of CMLP-CS andCM with oral administration, respectively
4 讨论
姜黄素是一类溶解性低、渗透性高的药物,其口服生物利用度低。本文采用经典的薄膜分散法制备CM纳米脂质体,可改善药物的溶解性,并在脂质体的表面包覆有天然多糖壳聚糖,其生物相容性好、黏附性强,能够打开胃肠道上皮细胞的膜孔,促进大分子穿过上皮组织的转运,并通过黏附作用延长脂质体在体内的滞留时间,从而提高生物利用度。
本文将CM制备成CMLP-CS后,在相同的释放介质中,48 h以前CMLP-CS的释放速率略快于CM,其原因可能是少部分未包封的CM与两亲性的磷脂结合快速释放,而48 h后,游离的CM的累积释放率已达到最高,药物释放已接近饱和,而CMLP-CS仍然持续释放药物至148 h,具有良好的缓释效果。其原因可能是包载在亲水中心的CM不仅需要穿过磷脂双分子层,还需要穿过脂质体表面的壳聚糖包衣从而进行释放,双重“屏障”可能是其缓释作用的主要原因。
在体内药代动力学研究中,CMLP-CS和CM的药时曲线均出现了双峰。CM的药时曲线出现双峰现象可能是由于CM经口服后在体内快速向组织分布,其中一部分在肝脾等器官中蓄积后再次释放入血。而CMLP-CS的药时曲线出现双峰的原因可能如下:(1)未包封的CM快速入血,包封在脂质体内的CM由于需要穿过磷脂和壳聚糖双重屏障而达到持续释放入血,从而达到最大血药浓度;(2)具有生物黏附性的壳聚糖增加药物与胃肠道接触的面积与时间,从而延长了药物的释放时间。药时曲线和主要药代参数均表明,CMLP-CS可明显提高CM的在体内的口服生物利用度,其表现出的缓效长效作用与体外释放动力学考察的结果一致。综上所述,CMLP-CS可增加体外释药量,改善体外释放行为,明显提高姜黄素的口服生物利用度,具有良好的促进吸收作用,为进一步研究姜黄素口服纳米制剂提供了理论基础。
[1] Wanninger S, Lorenz V, Subhan A, et al. Metal complexes of curcumin-synthetic strategies, structures and medicinal applications[J].ChemSocRev, 2015,15: 4986-5002.
[2] Zhang W, Chen C, Shi H, et al. Curcumin is a biologically active copper chelator with antitumor activity[J].Phytomedicine, 2016,23(1):1-8.
[3] Heger M, van Golen R F, Broekgaarden M, et al. The molecular basis for the pharmacokinetics and pharmacodynamics of curcumin and its metabolites in relation to cancer[J].PharmacolRev, 2014,66: 222-307.
[4] Patil S, Choudhary B, Rathore A, et al. Enhanced oral bioavailability and anticancer activity of novel curcumin loaded mixed micelles in human lung cancer cells[J].Phytomedicine, 2015,22(12):1103-11.
[5] 于梦迪,王明霞,王海东. 姜黄素逆转非小细胞肺癌分子靶向药物耐药的研究进展[J]. 中国药理学通报, 2017,33(12):1633-7.
[5] Yu M D, Wang M X, Wang H D, et al. Research progress of curcumin in reversing the drug resistance of non-small cell lung cancer[J].ChinPharmacolBull, 2017,33(12):1633-7.
[6] 李昕阳,谢 辉,严国俊. 壳聚糖衍生物在生物黏附药物传输系统中的研究进展[J]. 中国药学杂志, 2014,49(23):2053-7.
[6] Li X Y, Xie H, Yan G J. Progress in research of chitosan derivatives in bioadhesive drug delivery system[J].ChinPharmJ, 2014,49(23):2053-7.
[7] 廖 红,董 志,朱 毅,等. 壳聚糖对大鼠Ⅲ度烧伤创面成纤维细胞生物学行为的影响[J]. 中国药理学通报,2008,24(7):947-50.
[7] Liao H, Dong Z, Zhu Y, et al. Changes of the biological behavior of dermal fibroblasts in III skin burns wound in rats using chitosan[J].ChinPharmacolBull, 2008,24(7):947-50.
[8] 焦延鹏,李立华,罗丙红,等. 壳聚糖对骨组织工程中组织修复的影响[J]. 中国材料进展, 2012,31(9):35-9.
[8] Jiao Y P, Li L H, Luo B H, et aL. Effect of chitosan on tissue repair in bone tissue engineering[J].MaterialsChina, 2012,31(9):35-9.
[9] Muzzarelli R A, El Mehtedi M, Bottegoni C, et al. Genipin-crosslinked chitosan gels and scaffolds for tissue engineering and regeneration of cartilage and bone[J].MarDrugs, 2015,13(12):7314-38.
[10] 曾 梅,钟 萌,冯 悦,等. 阳离子阿奇霉素微类脂体体内外相关性[J]. 中国药理学通报, 2017,33(12):1713-7.
[10] Zeng M, Zhong M, Feng Y, et al.Invivoandinvitrocorrelation of azithromycin cationic micron niosomes[J].ChinPharmacolBull, 2017,33(12):1713-7.
[11] 袁誉铭,陈学梁,陈 静,等. 溴吡斯的明新型纳米乳体外释放和大鼠在体胃肠吸收[J]. 中国药理学通报, 2017,33(2):276-9.
[11] Yuan Y M, Chen X L, Chen J, et al. Preliminary research on drug releaseinvitroand intestinal absorption of pyridostigmine bromide nanoemulsion in the rats[J].ChinPharmacolBull, 2017,33(2):276-9.
[12] 赵 静,李 嫄,石明芯,等. 姜黄素乙醇脂质体大鼠体内药代动力学研究[J]. 四川大学学报(医学版), 2017,48(2):290-4.
[12] Zhao J, Li Y, Shi M X, et al. Pharmacokinetics of curcumin ethosomes in ratsinvivo[J].JSichuanUniv(MedSciEd), 2017,48(2):290-4.
[13] 陈贤春,吴 清,王玉蓉,等. 关于溶出曲线比较和评价方法[J]. 中国医院药学杂志, 2007,27(5):662-4.
[13] Chen X C, Wu Q, Wang Y R, et al. Comparison and evaluation of dissolution curve[J].ChinJHospPharm, 2007,27(5):662-4.
[14] 毕文杰,穆小静,祖丽皮艳·阿布力米特. 姜黄素新型靶向制剂研究进展[J]. 中国药学杂志, 2015,50(4):323-9.
[14] Bi W J, Mu X J, Abulimite ZLPY. Progress in targeting-delivery systems for curcumin[J].ChinPharmJ, 2015,50(4):323-9.
[15] Tan Q, Liu S, Chrn X, et al. Design and evaluation of a novel evodiamine-phospholipid complex for improved oral bioavailability[J].AAPSPharmSciTech, 2012,13(2): 534-47.
猜你喜欢
化工管理(2022年14期)2022-12-02
Digital Chinese Medicine(2022年2期)2022-07-02
九江学院学报(自然科学版)(2022年2期)2022-07-02
昆明医科大学学报(2022年3期)2022-04-19
药学研究(2021年5期)2021-11-29
昆明医科大学学报(2021年6期)2021-07-31
昆明医科大学学报(2021年3期)2021-07-22
信息技术时代·上旬刊(2019年4期)2019-09-10
天津医科大学学报(2019年3期)2019-08-13
Medical Data Mining(2019年2期)2019-07-16
