
湖南桃江病圃92个稻瘟病菌株遗传多样性分析
2018-06-05 05:57任佐华刘品克张译允王恒沪刘二明
西南农业学报 2018年4期
周 瑚,任佐华,刘品克,张译允,王恒沪,刘二明*
(1. 湖南农业大学植物保护学院,湖南 长沙 410128;2. 植物病虫害生物学与防控湖南省重点实验室,湖南 长沙 410128;3. 南方粮油作物协同创新中心,湖南 长沙 410128)
【研究意义】稻瘟病是由稻瘟病菌(有性世代Magnaportheoryzae,无性世代Pyriculariaoryzae)引起的一种世界性重要水稻真菌性病害[1],能侵染水稻组织的各个生育期,并可以寄生在小麦、大麦和粟等多种禾本科作物及杂草上[2-3]。目前,稻瘟病的防治以选育及推广利用抗病品种为核心,以健康、合理搭配栽培等农业措施为基础,适当利用化学农药防治为辅助的综合策略[4];但稻瘟病菌非常复杂易变,而且水稻品种在推广种植3~5年后抗性表现易“丧失”。【前人研究进展】自20世纪80年代末以来,随着分子生物学的发展,以DNA多态性分析为基础的分子标记技术已经被广泛用于稻瘟病菌的群体分析,DNA多态性指的是生物之间在DNA水平上的差异,它为植物病原真菌的种类鉴定提供了大量的遗传标记[5]。利用RFLP(限制性片段长度多态性,Restriction Fragment Length Polymorphism)、RAPD(随机扩增多态性DNA,Random Amplified Polymorphic DNA)、AFLP(扩增片段长度多态性,Amplified Fragment Length Polymorphism)、SCAR(Sequence Characterized Amplified Region)、ISSR(inter-simple sequence repeat)和SSR(简单重复序列,Simple Sequence Repeat,SSR)等技术对稻瘟病菌进行分析,研究其遗传规律。【本研究切入点】SSR分子标记技术因具有易检测、多态性高、可重复性高、结果稳定等优点[6],因而成为分析稻瘟病菌遗传多样性的主要方法。【拟解决的关键问题】为解决水稻品种在推广种植抗性表现易“丧失”、呈“感病化”的问题,本研究利用SSR技术对2015年分离于湖南桃江病圃的92个稻瘟病菌菌株进行遗传多样性分析,为湖南省水稻抗瘟性监测提供依据。
1 材料与方法
1.1 供试菌株
2015年6-7月在湖南省益阳市桃江县稻瘟病圃(N28°22′, E112°03′)内采集普感稻瘟病品种“LTH”的叶瘟病样,参照并改良张建书等[7]的挑取单孢方法,从病标上共分离保存92个稻瘟病单孢菌株。
1.2 供试引物
由上海生工公司合成的14对SSR引物(表1),分别为KMS02、KMS20、SMS17、A5、D4、D5、G5、FG01、FG02、FG03、MS355、MS363、MS677[8]、PYRM419[9]。
1.3 主要仪器
恒温光照培养箱(GZX-250BS-Ⅲ,上海新苗医疗器械制造有限公司)、显微镜(北京普瑞赛司仪器有限公司)、恒温摇床(Crystal)、高速冷冻离心机(Eppendorf)、震荡涡旋仪(海门市其林贝尔仪器制造有限公司)、恒温水浴锅(HH-6,上海浦东物理光学仪器厂)、微量分光光度计(BIOSPEC-NANO)、PCR扩增仪(联想生物科技有限公司)、水平电泳仪(北京六一仪器厂)、紫外凝胶成像系统仪(BIO-PRO 200E)。
1.4 主要试剂及培养基
DNA提取试剂盒(EasyPure Genomi DNA Kit),2K DNA Marker,SYBR Green I染料,6×DNA Loading Buffer,山梨醇Buffer,(购自北京全式金生物技术有限责任公司,北京);TBE缓冲液;可溶性淀粉培养基;液体培养基[10]。
1.5 稻瘟病菌丝的培养
将用高粱粒保存的稻瘟病菌接种到可溶性淀粉平板培养基中,26 ℃,光暗交替(L/D=12h/12h)培养5 d左右,从最外缘刮去带少量培养基的菌丝接种到盛有40 mL液体培养基的三角瓶中,28 ℃,180 r/min恒温摇床培养3~5 d。
1.6 DNA提取及浓度纯度测定
用试剂盒法(北京)提取稻瘟病菌基因组DNA。将提取的DNA使用Nano Drop的Spectrophotometer检测样品DNA的OD260、OD280的值,通过OD260/OD280检测DNA浓度(纯DNA:OD260/OD280≈1.8,>1.9,表明有RNA污染;<1.6表明有蛋白质、酚等污染),并调整DNA纯度为150 ng/μl。

表1 供试引物
1.7 PCR扩增
PCR扩增体系见表2。PCR扩增程序见表3。
1.8 琼脂糖凝胶电泳
利用质量体积分数为2 %的琼脂糖凝胶电泳检测PCR扩增产物。第1孔加入由3 μl Green I染料和7 μl Marker制成的混合液8 μl;第2孔加由3 μl染料、2 μl Loading Buffer和5 μl ddH2O制成的混合液8 μl;第3~25孔加入由3 μl 染料、2 μl Loading Buffer及5 μl PCR产物混合而成的上样液8 μl。在110 V电压强度下,35 min,条带跑至胶的1/2~2/3处即可。电泳完成后在凝胶成像系统下观察、拍照记录。
1.9 数据处理
根据SSR引物扩增电泳结果,将电泳图片转换为2进制的数据(有条带记的为1,无条带的记为0),应用软件DPS 7.05 Nei & Li最长距离法处理数据得到聚类分析图。
2 结果与分析
2.1 SSR引物对供试菌株的PCR扩增
92个供试菌株能与14对用于分析的SSR引物特异性结合并有效扩增,所得重复序列因引物不同而不同,电泳检测后能较清晰表现出扩增的多态性。KMS02、KMS20、SMS17、A5、D4、D5、G5、FG01、FG02、FG03、MS355、MS363、MS677、PYRM419分别对55、46、47、44、42、44、49、78、77、70、59、64、59、63个菌株扩增出特异性亮带,Marker条带重复性良好,CK无条带出现(图1)。

表2 PCR扩增体系
2.2 病圃稻瘟病菌群体遗传谱系
利用14对SSR引物对92个稻瘟病菌单孢菌株进行PCR扩增后的电泳结果转换为0-1数据,通过数据分析软件DPS 7.05进行聚类分析,结果(图1,表4)表明,在相似系数0.19,即差异系数0.81的水平,92个供试的稻瘟病菌单孢菌株被划分在同一个宗谱内;在相似系数0.70,即差异系数0.30的水平,供试的92个菌株被划分为26个宗谱,统计各宗谱内的菌株数可知XIX宗谱为优势宗谱,含有17个菌株,占总菌株数的18.48 %;IV宗谱中含有10个菌株,占总菌株数的10.87 %;XVIII宗谱中含有9个菌株,占总菌株数的9.78 %;其他23个宗谱内各含1~5个菌株,共占总菌株数的60.87 %。从聚类分析图谱看来,从湖南桃江病圃分离的稻瘟病菌遗传差异较大,遗传结构比较复杂,个别菌株亲缘关系较远。
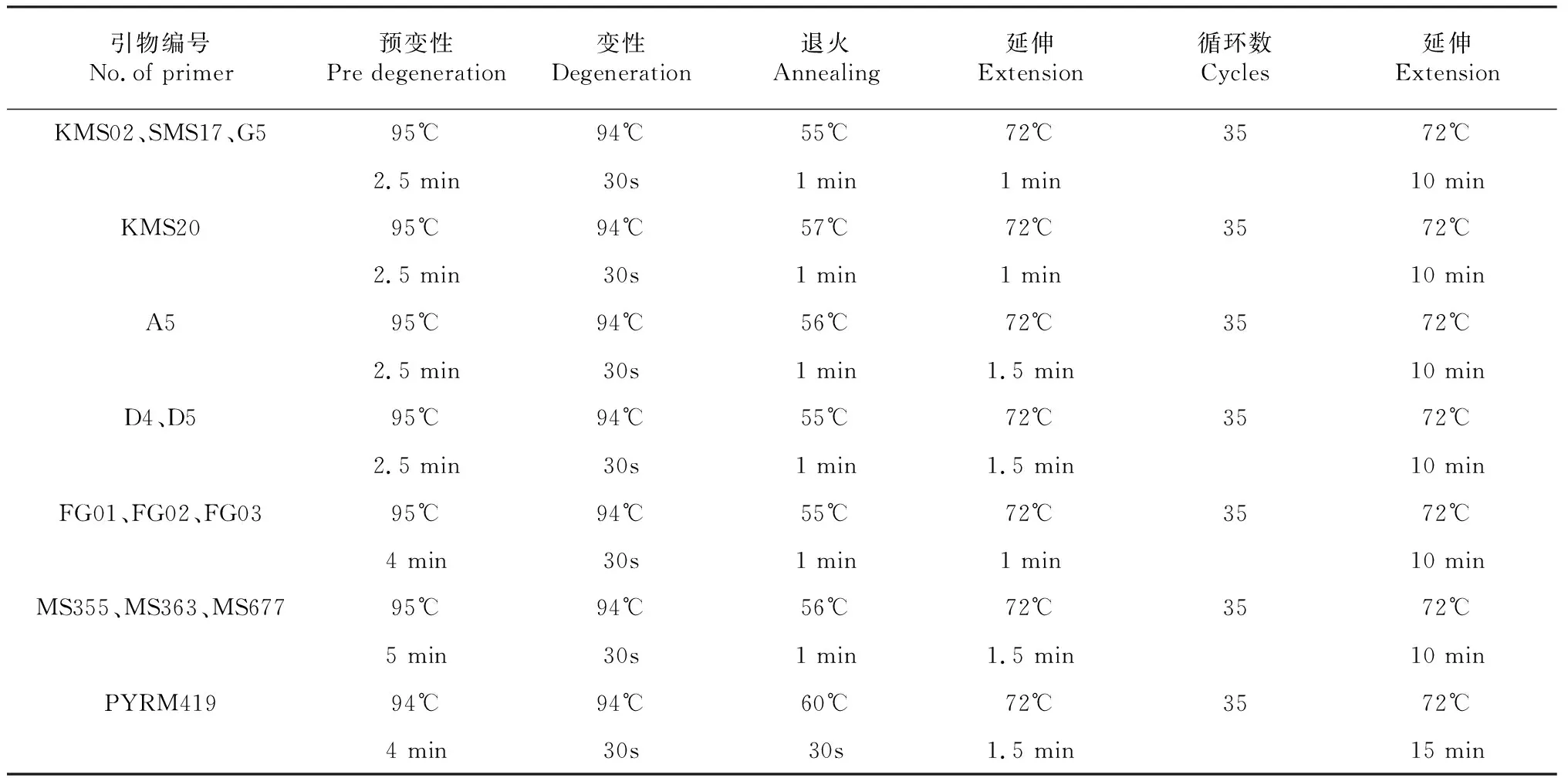
表3 PCR反应程序
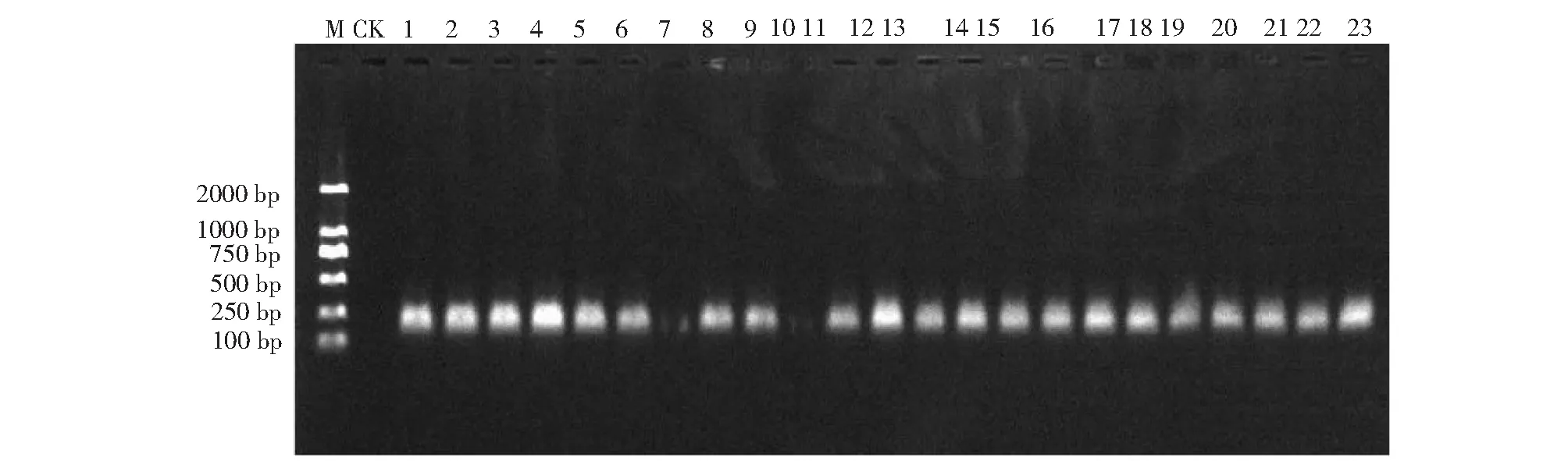
M:已知分子量的DNA混合物;CK:ddH2O;1~23:供试菌株M: DNA Markerr; CK: ddH2O; 1-23:Tested strains图1 引物FG03对部分菌株的扩增结果Fig.1 The amplification results of some texted strains for primer FG03
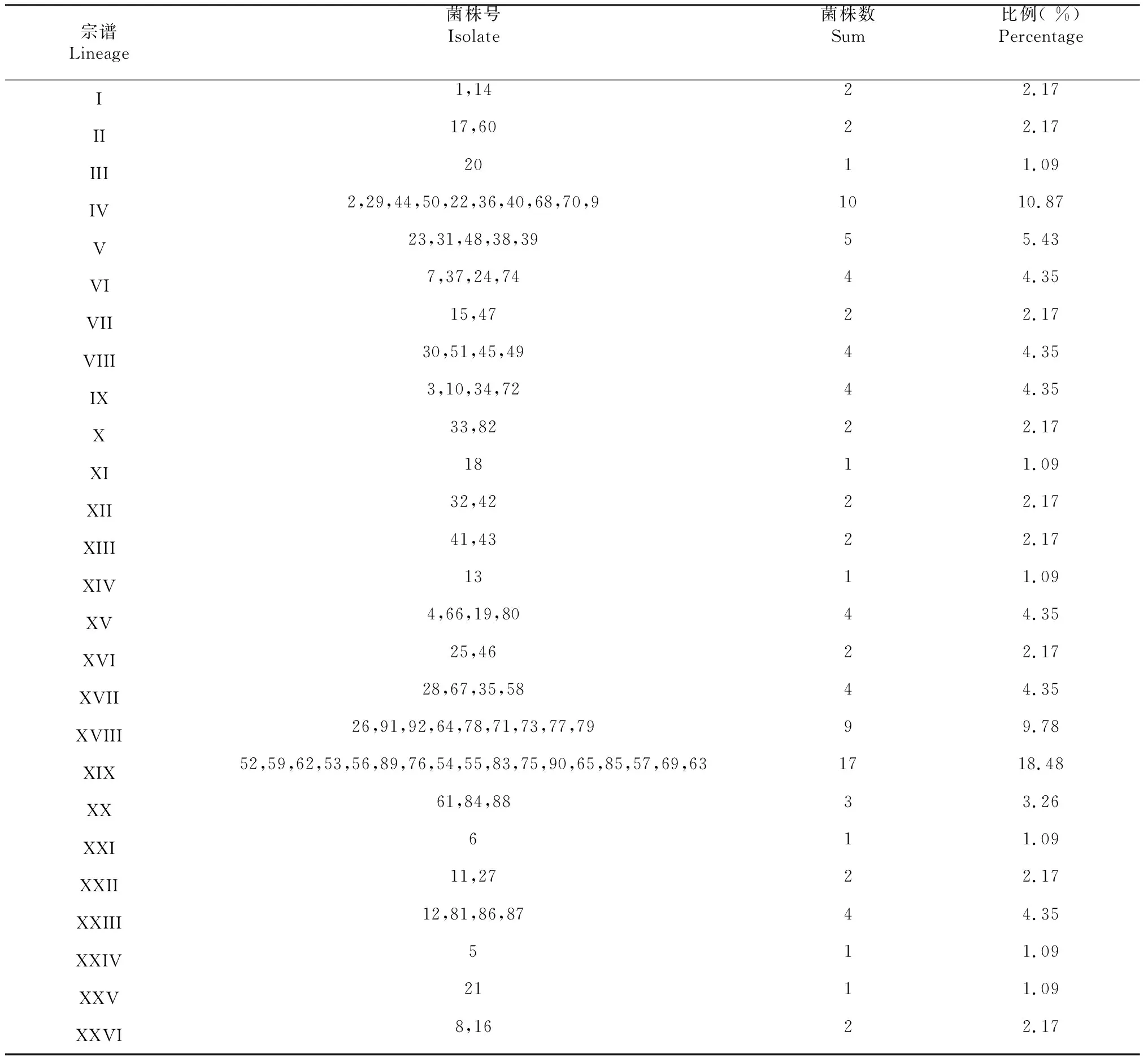
宗谱Lineage菌株号Isolate菌株数Sum比例( %)PercentageI1,1422.17II17,6022.17III2011.09IV2,29,44,50,22,36,40,68,70,91010.87V23,31,48,38,3955.43VI7,37,24,7444.35VII15,4722.17VIII30,51,45,4944.35IX3,10,34,7244.35X33,8222.17XI1811.09XII32,4222.17XIII41,4322.17XIV1311.09XV4,66,19,8044.35XVI25,4622.17XVII28,67,35,5844.35XVIII26,91,92,64,78,71,73,77,7999.78XIX52,59,62,53,56,89,76,54,55,83,75,90,65,85,57,69,631718.48XX61,84,8833.26XXI611.09XXII11,2722.17XXIII12,81,86,8744.35XXIV511.09XXV2111.09XXVI8,1622.17
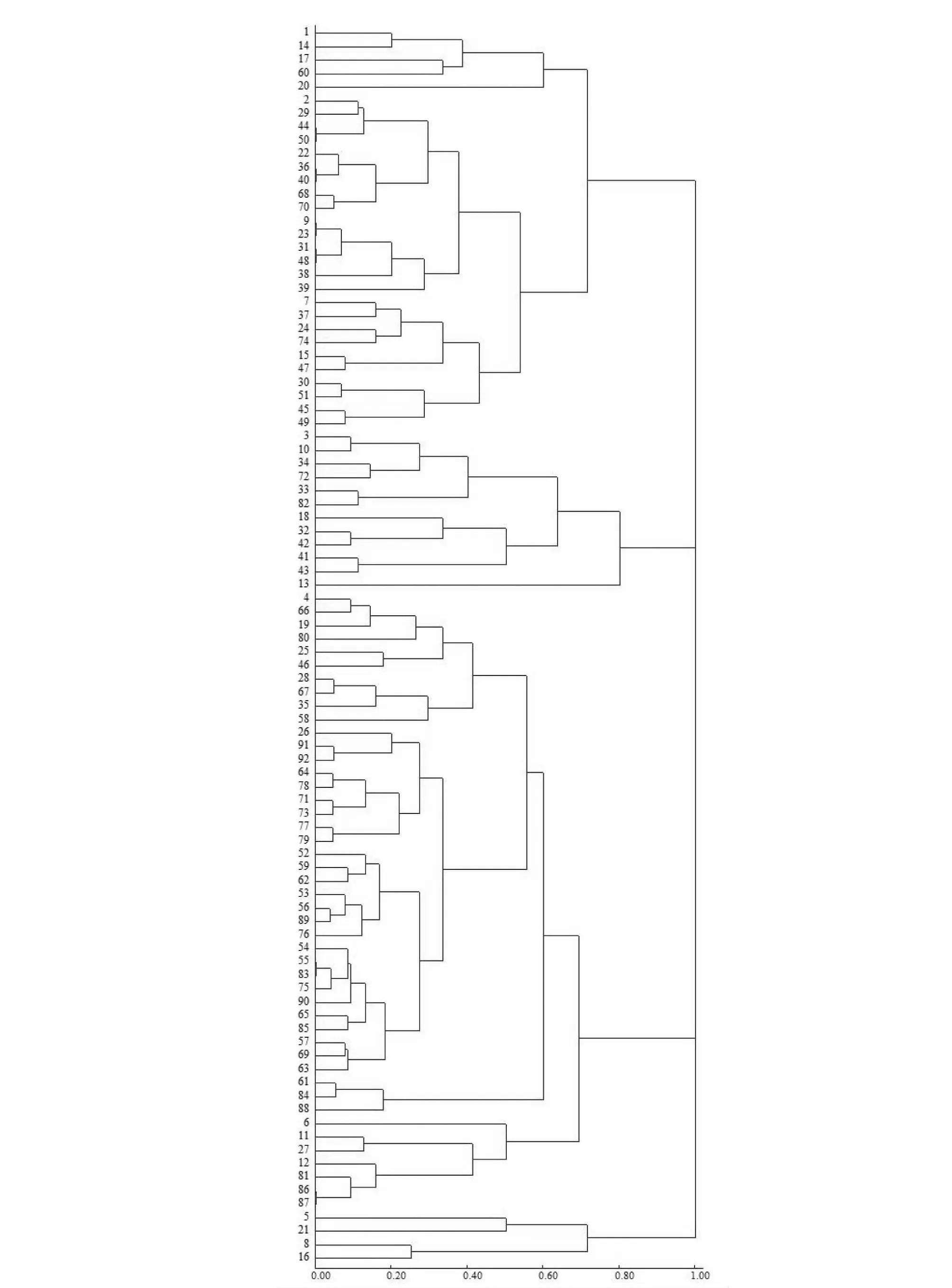
纵坐标1~92为试验菌株编号,横坐标0.00~1.00为相似距离The number of Magnaporthe oryzae on the vertical and correlation distance on the horizontal图2 92个稻瘟病菌菌株亲缘关系树状图Fig.2 Dendrograph constructed based on SSR primers indicating genetic relationship among 92 M. oryzae
3 讨 论
随着分子标记技术的快速发展,SSR标记被广泛应用于稻瘟病菌种群结构的遗传多样性分析,Zheng等(2000)[11]设计并筛选了446对SSR引物,其中313对引物具有较高的多态性;Brondani等(2008)[12]设计了24对SSR引物,用于稻瘟病菌遗传多样性的研究;毛建辉(2008)[13]对四川45个菌株进行遗传多样性分析,研究表明在0.76相似水平上可被划分为7个宗谱,优势宗谱内的菌株所占比列为71.11 %;周燕(2016)[14]对黑龙江省20个地区181个菌株进行遗传多样性分析,研究表明供试所有菌株在0.52相似水平上即可归纳在1个宗谱内,在0.72相似水平上划分成11个宗谱,其中7个宗谱内的菌株为1~3株。
刘二明(2002)[15]对湖南129个菌株的稻瘟病菌菌株进行遗传多样性分析,研究表明在0.72相似水平被划分成4个遗传宗谱,优势宗谱内菌株所占比例为65 %,揭示了稻瘟病菌存在较大的变异潜能;李亚(2007)[16]对湖南230个菌株稻瘟病菌菌株进行遗传多样性分析,表明在0.77相似水平上可被划分成7个遗传宗谱,其中Ⅳ和Ⅶ宗谱中分别只有1个和5个菌株;童建新(2012)[17]对湖南19个县市的169个菌株进行遗传多样性分析,结果表明在0.80相似水平上可被划分成8个遗传宗谱,优势宗谱内菌株所占比例为66.86 %;毛锐(2015)[18]对湖南桃江病圃89个菌株进行遗传多样性分析,结果表明在0.72相似水平上可被划分成20个遗传宗谱,优势宗谱内菌株所占比例为20.20 %;刘翔(2016)[19]对湖南桃江病圃120个菌株进行遗传多样性分析,结果表明在0.75相似水平上可被划分成32个遗传宗谱,优势宗谱内菌株所占比例为16.67 %,其中27个宗谱内的菌株为1~6株。
4 结 论
LTH是中国稻瘟病菌生理小种的鉴别品种之一,具有普感稻瘟病的特性。本研究所用的菌株均分离自LTH的叶瘟病样,这使得稻瘟病菌群体能更真实地反应稻瘟病菌自然种群的遗传结构;湖南桃江病圃是国家水稻区域试验长江中游生态区抗稻瘟病鉴定基地,每年为全国科研、生产单位鉴定大量育种材料和生产品种,比较而言,该病圃的病菌群体遗传结构比其它生态系的稻瘟病菌复杂,周益军等[20]认为水稻品种越丰富,稻瘟病菌的遗传复杂性和多样性的程度越高。本研究利用14对SSR引物对分离自桃江LTH上的92个菌株进行遗传多样性分析,结果表明在0.70相似水平的92个供试菌株共被划分为27个宗谱,综合聚类分析图谱看来,92个供试稻瘟病菌遗传差异较大,个别菌株亲缘关系较远,遗传结构比较复杂;源自同一水稻品种的稻瘟病菌遗传多样性也较为丰富,这进一步了证明地域品种的多样化,导致稻瘟病菌群体遗传的多样性。
参考文献:
[1]Skamnioti P, Gurr S J. Against the grain: safeguarding rice from blast disease [J]. Trends in Biotechnology, 2009, 27(3):141-150.
[2]Tosa Y, Chuma I. Classification and parasitic specialization of blast fungi[J]. Journal of General Plant Pathology, 2014, 80(3):202-209.
[3]Zeigler R S. Recombination in magnaporthe grisea[J]. Annual Review of Phytopathology, 1998, 36(36):249-275.
[4]Yang S, Li J, Zhang X, et al. Rapidly evolving R genes in diverse grass species confer resistance to rice blast disease[J]. Proceedings of the National Academy of Sciences of the United States of America, 2013, 110(46):18572-7.
[5]薛文君,黄 敏,卢代华,等. 稻瘟病菌多样性研究进展[J]. 西南农业学报, 2007, 20(1):157-162.
[6]林富荣,邢俊连,孟艳琼,等. 皂荚EST-SSR分子标记开发与分析评价[J]. 植物遗传资源学报, 2017, 18(1):148-154.
[7]张书建,何月秋. 介绍一种简单的真菌单孢子分离法[J]. 云南农业大学学报自然科学, 2003, 18(3):315-316.
[8]虞选杰,任佐华,管玲莉,等. 湖南桃江病圃丽江新团黑谷上稻瘟病菌遗传多样性分析[J]. 杂交水稻, 2014, 29(3):70-73.
[9]谢晶晶,姜 华,毛雪琴,等. 利用SSR技术研究浙江省稻瘟病菌群体的遗传多样性[J]. 浙江农业学报, 2015, 27(10):1781-1788.
[10]刘 翔,任佐华,陈娟芳,等. 湖南省稻瘟病菌无毒基因鉴定[J]. 南方农业学报, 2016, 47(9):1500-1505.
[11]Zheng Y, Zhang G, Lin F, et al. Development of microsatellite markers and construction of genetic map in rice blast pathogen Magnaporthe grisea[J]. Fungal Genetics & Biology Fg & B, 2008, 45(10):1340-1347.
[12]Brondani C, Brondani RPV, Garrido LDR, et al. Development of microsatellite markers for the genetic analysis of Magnaporthe grisea.[J]. Genetics & Molecular Biology, 2000, 23(4):753-762.
[13]毛建辉,王 平,卢代华,等. 利用SSR技术分析混栽净栽水稻田间稻瘟病菌的遗传多样性[J]. 西南农业学报, 2008, 21(5):1294-1297.
[14]周 燕. 黑龙江省稻瘟病菌培养性状和致病性及遗传多样性的分析[D]. 黑龙江: 黑龙江八一农垦大学硕士学位论文, 2015.
[15]刘二明,张志飞,罗 峰,等. 湖南稻瘟病病菌群体遗传多样性研究[J]. 湖南农业大学学报(自科版), 2002, 28(5):391-394.
[16]李 亚,刘二明,戴良英,等. 湖南稻瘟病菌群体遗传多样性与病菌致病型的关系[J]. 中国水稻科学, 2007, 21(3):304-308.
[17]童建新,任佐华,刘毅雄,等. 湖南稻瘟病菌遗传多样性分析[J]. 杂交水稻, 2012, 27(3):66-70.
[18]毛 锐. 湖南桃江病圃稻瘟病菌遗传多样性及病圃无毒基因和主栽水稻品种抗瘟基因型鉴定[D]. 长沙: 湖南农业大学硕士学位论文, 2014.
[19]刘 翔,任佐华,陈娟芳,等. 利用SSR分析湖南桃江病圃丽江新团黑谷上稻瘟病菌的遗传多样性[J]. 西南农业学报, 2015, 28(6): 2496-2500.
[20]周益军,白 娟,程兆榜,等. 我国稻瘟病菌群体多样性研究[J]. 中国水稻科学, 2004, 18(3):277-280.
猜你喜欢
智慧农业导刊(2022年21期)2022-11-03
作物学报(2022年6期)2022-04-08
文物鉴定与鉴赏(2022年2期)2022-03-30
文萃报·周五版(2021年25期)2021-08-06
农民致富之友(2020年15期)2020-05-25
农民文摘(2019年7期)2019-01-14
农业与技术(2018年8期)2018-06-21
辽河(2015年2期)2016-04-06
黑龙江史志(2013年3期)2013-08-15
寻根(2009年6期)2009-05-14
