
抗生素耐药基因污染研究进展
2018-06-01 01:54武中庸赵吴静蒲万霞
中国草食动物科学 2018年3期
武中庸,赵吴静,侯 晓,蒲万霞
(中国农业科学院兰州畜牧与兽药研究所,中国农业科学院新兽药工程重点开放实验室,甘肃省新兽药工程重点实验室,兰州 730050)
抗生素耐药性与抗生素的使用相生相伴。某些环境微生物本身携带或者能够从其他菌株获得某些特定基因,这些基因能够通过编码具有不同功能的蛋白质以达到去除抗生素效应的目的,其被称为抗生素耐药基因(Antibiotic resistance genes,ARGs)[1]。Rysz 等于 2004 年将ARGs归纳为一类新型环境污染物,Pruden等于2006年正式明确提出这一概念,从此ARGs愈受关注。ARGs可分为“内源性耐药基因”和“外源性耐药基因”。内源性耐药基因主要指本身存在于基因组上的耐药基因原型、准耐药基因及平时未表达的耐药基因,其能够随机突变或通过外部选择压力表达耐药性。D’Costa等[2]从北极3万年冻土中发现,对β-内酰胺类、四环素类和糖肽类等抗生素具有抗性的ARGs,且万古霉素耐药蛋白结构和功能与现代变体极为相似,这表明某些类型ARGs早就存于自然界,而不是现代使用抗生素造成的。外源性耐药基因主要指ARGs在外在压力下选择性迁移到敏感菌中,从而使得敏感菌获得耐药性。
1 ARGs多重抗性的形成
ARGs可随抗性细菌从粪便进入环境,并通过水平转移机制(Horizontal gene transfer,HGT)进入各种环境土著菌,其扩散效率可能超过亲代菌株[3];同时,携带ARGs的裸露DNA可在菌株死亡后释放进入环境中且长期存在。畜禽养殖因为产业的特殊性,抗生素的大量使用不可避免,因此也极大地刺激了ARGs的发展,给予了ARGs发展的外在选择压力。同时,因为畜禽养殖人员关于抗生素污染的意识单薄和畜禽粪便处理措施匮乏,大部分畜禽粪便未经处理或者只经过简单处理就进入环境,尤其是土壤环境;动物肠道内和粪便中含有丰富的抗性细菌和ARGs,因此进入农田土壤、水源、地下水等周边环境,极大地增加了周边环境的ARGs丰度及土著耐药菌的产生。未经处理的畜禽粪便大量输入环境,对环境中细菌的耐药水平起到了重要的作用,其可诱导多重耐药基因在细菌中组合,细菌呈多重耐药性。目前,细菌多重耐药现象已引起各方日益关注。典型的多重耐药致病菌包括:泛耐药鲍氏不动杆菌(Pandrugresistant Acinetobacter baumannii,PRAB)、耐甲氧西林金黄色葡萄球菌(Methicillin-resistant Staphylococcus aureus,MRSA)、碳青霉烯类耐药铜绿假单胞菌(Carbapenem-resistant Pseudomonas aeruginosa,CRPA)、携NDM-1(New Delhi metallo-β-lactamase-1)耐药基因菌、质粒介导的黏菌素耐药基因mcr-1等[4]。细菌中的多重耐药基因常以耐药基因盒或基因岛的方式存在。中国学者发现,猪源 Escherichia coli的Ⅰ型整合子上 aadA1、blaP1a-aadA2-ereA、aadA2、dfrA1-aacA4-catB3、aadA23B、dfrA1-orfC和dfrA1-aadA1等耐药基因均存在于耐药基因岛上[5]。Neyra 等[6]研究发现,金黄色葡萄球菌对甲氧西林的耐药率14.3%,其多重耐药率37.1%。
2 ARGs分类
根据抗生素抑菌机制的不同可以将抗生素分为β-内酰胺类、四环素类、氨基糖苷类、磺胺类、糖肽类、喹诺酮类、氯霉素类、大环内脂类等大类,ARGs也可以根据抗生素类别的不同进行分类,其分类情况详见表1。
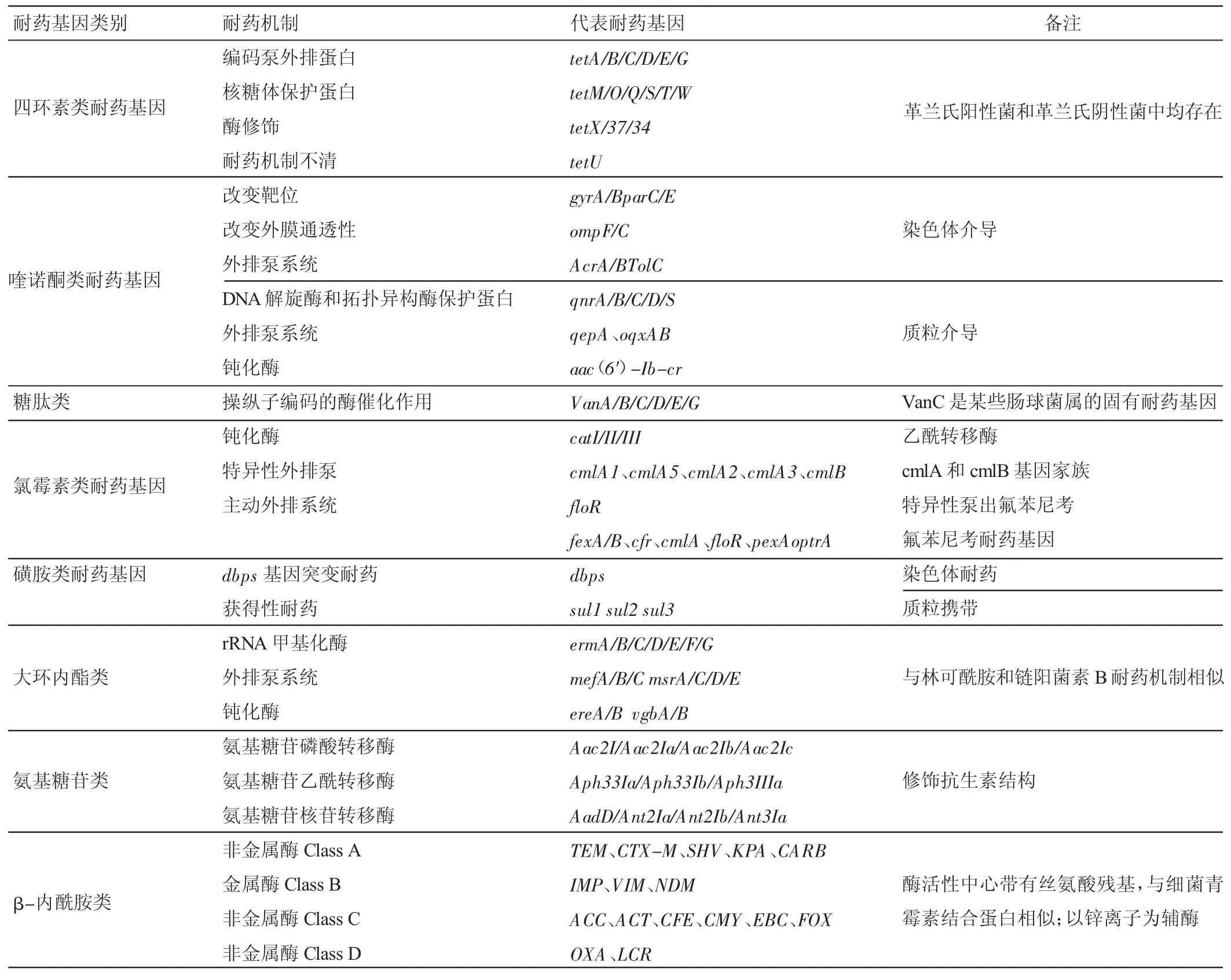
表1 不同抗生素类别耐药基因
3 ARGs研究进展
ARGs的形成与传播,是细菌产生耐药性的内在因素。随着科技的发展和研究手段的不断优化,人们对细菌染色体和基因组的研究越发深入,各种ARGs不断被发现,使人们对细菌耐药性的形成机制也有了新的认识。因ARGs传播和调控过程在细菌基因组内的复杂性,借由对ARGs的研究,对细菌基因组的研究也得以拓展和深入,而细菌基因组研究也会大大促进了ARGs的研究进程。
3.1 传统ARGs的研究方法
传统ARGs的研究方法主要为微生物培养鉴定方法,其主要步骤为:①目的样品采集;②对目的样品中细菌进行分离纯化鉴定,常采用适应不同细菌生长的培养基进行菌株分离纯化,以生化鉴定方法或者16S rRNA方法对菌株进行种群鉴定,以尽量获取培养菌株信息;③对分离细菌进行抗生素敏感性实验,以获取菌株耐药表型,抗生素敏感性实验经常测定菌株的药物最低抑菌浓度(Minimal inhibitory concentration,MIC),通过MIC判定菌株的耐药表型。得益于生物技术的飞速发展,ARGs的研究方法也取得了巨大进步。聚合酶链式反应(Polymerase chain reaction,PCR)是一种能够根据已知目的基因进行保守序列的引物设计,并以DNA合成基本原料,经变性、退火、延伸过程在生物体外快速扩增特定DNA片段的分子生物学技术。但是,常规PCR扩增方法具有较大缺点,其只能定性检测耐药基因存在与否。实时荧光定量PCR(Real-time quantitative PCR,RTQ-PCR)应运而生,有效解决了常规PCR扩增方法只能定性检测基因的问题。该方法通过在PCR反应体系中加入荧光基团,以积累的荧光信号的量来实时对PCR进程进行监测,最后与标准曲线对比进行耐药基因丰度的检测。但是,普通PCR和RTQ-PCR均局限于已知序列耐药基因检测,其不能用于新基因的发现;且如需进行大量基因检测时具有较大的工作量。Popowska等[7]采用定量PCR研究了农业土壤中四环素类、氨基糖苷类类和大环内酯类耐药基因的分布特征。
3.2 基因芯片检测技术
20世纪80年代末发展起来的基因芯片技术克服了PCR方法样品检测量小的缺点,其采用集约化和平面处理,在固相载体上固定了数以百万计的DNA探针,通过将样品与芯片探针杂交来检测样品中DNA序列。该方法适用于大量样品的检测并具有减小工作量和降低用时的特点。基因芯片技术主要从两方面实现耐药基因的检测:①借助寡核苷酸芯片检测基因组序列的亚型或突变位点;②以基因表达谱芯片检测药物诱导基因表达的改变[8]。Bruant等[9]设计了检测大肠杆菌毒力基因和耐药基因的基因芯片,其可检测致病菌株及其耐药基因。基因芯片可用于研究环境微生物的群落结构、代谢途径、追踪关键基因等方面。但是基因芯片也有一系列问题需要解决,如:费用过高,不合适普通临床检测;基因序列库尚小,需加强建库工作;基因芯片专利限制,不同公司专利权不同,需加强行业信息整合。
3.3 宏基因检测方法
微生物纯化培养方法对于研究细菌的耐药性起到了重要作用,但传统微生物培养法具有一定的局限性。澳大利亚学者Heinrich Winterberg首先观察到细胞计数方法和平板计数方法得到的样品中微生物总量存在巨大差异,因为存在于环境中的微生物绝大部分是不可培养的,传统微生物培养方法能培养的微生物仅占环境中微生物总量不到1%,环境中尚有大量的耐药微生物被遗漏。
Pace于1985年第一次提出可以从环境中直接提取DNA来研究不可培养微生物的理念。1995年,Healy等对宏基因文库成功地进行了功能筛选,这为后续概念提出和实验完善提供了基础。1998年,Handelsman等[10]正式提出了宏基因组学(Metageomics)的概念,即从环境中提取总DNA中用于进行遗传组成和功能研究。宏基因组学研究的发展跨越了传统微生物培养研究的瓶颈,让人们重新认识了环境中微生物群落的多样性,极大地拓展了微生物学研究的思路和方法。宏基因组学研究微生物的主要方法包括3个过程:①环境样品总DNA提取。DNA提取过程需根据样品的不同特点选择不同的提取方法,而不同的提取方法得到的提取结果也会有所差异,这也直接影响了后续试验[11]。②建立宏基因文库并测序。测序时可通过不同专用平台进行测序,但不同测序平台得到的最终结果有所差异,目前常用的测序平台有Illumina MiSeq和Illumina HiSeq平台。③序列拼接与分析。理论上选择合适的样品总DNA提取方法并测得一定的测序深度后获得的信息能够全面反映样品的遗传信息,获得微生物全基因组信息。宏基因组学主要分为功能宏基因组学和序列宏基因组学。可以将未知耐药基因的鉴定归为功能宏基因组学方向。目前,宏基因测序在环境菌群分析、肠道菌群分析、耐药基因检测鉴定方面发挥了重要作用。耐药基因相关比对数据库有:SEED database“Resistance to antibiotics and toxic compounds”、Antibiotic Resistance Genes Database(http://ardb.cbcb.umd.edu/)和 MetaGeneMark。
基于测序技术的飞速发展,尤其是二代测序应用以来,宏基因组方法在环境细菌耐药性上的应用研究得到了充分体现[12]。Chambers 等[13]通过宏基因组学测序方法发现,奶牛粪便中的β-内酰胺类耐药基因因为头孢类抗生素的使用而得到富集。有学者通过大量宏基因测序数据来比较分析不同来源ARGs的组成,确定了目前已知大部分ARGs在不同环境的分布规律[14]。Fang等[15]发现,蔬菜地土壤施用鸡粪后能使耐药菌和耐药基因富集,其检测到22大类ARGs和46种携带耐药基因的潜在致病菌。
3.4 微生物单细胞基因组技术
微生物单细胞基因组学是在宏基因组学后发展起来的能够有效获取无法人工培养微生物基因信息的新技术[16]。目前,环境微生物研究中主要将其应用于探索未被常规技术或宏基因组技术探测到的功能基因、发现丰度极小的未培养微生物或者研究微生物细胞生命进化过程等[17]。微生物单细胞基因组技术包括单细胞获取、全基因组扩增、全基因组测序以及数据分析等步骤。微生物单细胞获取常用的方法主要有:有限稀释、流式细胞分选、光镊抓取、显微操作和微流控芯片等,各种方法均有各自的优缺点,具体使用时需按实验目的选择。单个微生物细胞内含有很少量的DNA,需要使用特殊扩增方式将单细胞基因组从飞克级别扩增到纳克至微克级别才可用于后续分析[18]。Rinke 等[19]对 9 600 个环境微生物单细胞进行全基因组扩增和部分细胞测序,获得了201个未培养的细菌细胞及古菌,揭示了微生物之间的生物关系及其潜在代谢能力,并发现了新的嘌呤合成途径。微生物单细胞基因组技术在探测新型微生物或基因方面具有巨大的潜力,但是因为技术发展原因,该技术使用成本高昂,不适用于常规检测。
3.5 ARGs消除方法
目前,抗生素滥用导致的环境耐药菌增多,ARGs的转移已经成为不争的事实,由此所引发的生态环境问题和人员健康问题也不断得到人们关注。作为一个农业大国,我国畜禽产业快速发展,畜禽粪便产量连创新高,随粪便排出的抗生素量和由此诱导产生的抗性基因对环境造成的污染也越来越严重。为了降低环境中抗生素残留水平以及遏制抗性基因的传播扩散,必须采取有力措施,防止抗生素和抗性基因污染的进一步蔓延和恶化。3.5.1抗生素降解方法 抗生素自发现以来对控制人类疾病发挥了重要的作用,其同时也被用于畜禽养殖业,为人类畜产养殖业的发展做出了巨大贡献。然而,抗生素在全球范围内的滥用情况也十分严重,同时因其只有少量部分能被机体吸收,大部分通过代谢途径排出体外进入环境,由此引发的一系列环境问题及最终的对人类健康的影响问题也越来越受到人们的关注。加强抗生素降解方式的研究迫在眉睫。
自然环境是抗生素的受纳体,同时也是抗生素降解的促进体,但不同环境对于抗生素的降解作用有所不同[20]。光照是促进抗生素降解作用的重要因素。有研究表明,充足光照条件下,3 h内四环素、红霉素和土霉素的降解率分别可以达到66.87%、92.80%和90.55%[21]。有些抗生素本身为水溶性,极易溶于水,这在一定条件下也促进了抗生素的水解。在适宜条件(25℃,pH=7)下,青霉素、头孢西丁和头孢噻吩的半衰期在5.3~27 d范围内[22]。微生物降解也是去除环境中残留抗生素的有效方式,如添加选择性有益降解菌剂对于去除四环素类抗生素残留有很大效果[23]。堆肥处理是一种去除畜禽粪便中抗生素的有效手段。有学者研究发现,堆肥处理能够去除粪便中残留抗生素的50%~70%[24]。堆肥处理过程中处理好温度和氧气条件有利于更好地去除粪便中抗生素残留[25]。Arikan 等[26]发现,55℃条件下堆肥处理能够去除99%的金霉素,但是无温控堆肥仅有49%的去除率。
3.5.2 ARGs去除方法 ARGs去除与抗生素降解过程有一定相似之处,光照、温度和微生物群落结构等均可以影响ARGs的去除。有研究表明,ARGs降解过程可以因为光照环境而加速[27]。ARGs存在于微生物细胞内,不同微生物对于ARGs具有一定的耐受性,因此微生物群落的组成能够在一定程度上影响ARGs的含量,微生物菌群类型能够促使ARGs发生变化[28]。厌氧消化和好氧堆肥是畜禽粪便处理的主要工艺,这两种方式均对ARGs的去除具有良好的效果。Cheng等[29]对养猪场内不同工艺处理的废水进行了ARGs去除情况研究,发现厌氧消化处理能够降低大部分ARGs含量,但其他处理方法影响不大。利用堆肥过程产生的高温,可有效去除耐药质粒和 ARGs含量[30]。郑宁国等[31]研究了高温堆肥对猪粪来源ARGs的影响,结果表明,高温堆肥对β-内酰胺类ARGs和喹诺酮类ARGs具有一定去除能力,但大环内酯类ARGs去除效果不明显。
4 结语
抗生素滥用造成的环境污染问题及ARGs在菌群之间的转移引起的生态问题已引起各国学者和民众的高度关注。但环境中抗生素和ARGs的产生与存留是长期积累的过程,且在短期内不会消失。研究抗生素和ARGs的分布和行为特征等对人类采取合理对策减轻环境污染具有十分重要的意义。目前该领域的研究方向主要集中在对环境中抗生素和ARGs的监测,对于环境中ARGs存留及其影响因素、传递机制及其影响因素、迁移转化、人工去除等方面研究较少,未来针对ARGs研究建议从ARGs来源、传递机制、环境消除规律、迁移规律等方面进行。
[1]Martínez J L,Coque T M,Baquero F.What is a resistance gene?Ranking risk in resistomes [J].Nat Rev Microbiol,2015,13(2):116-123.
[2]D'Costa V M,King C E,Kalan L,et al.Antibiotic resistance is ancient[J].Nature,2011,477:457-461
[3]Chee-Sanford J C,Mackie R I,Koike S,et al.Fate and transport of antibiotic residues and antibiotic resistance genes following land application of manure waste [J].J Environ Qual,2009,38(3):1086-1108.
[4]Liu Yiyun,Wang Yang,Walsh T R,et al.Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China:a microbiological and lar biological study[J].Lancet Infect Dis,2016,6(2):161-168.
[5]Stokes H W,Gillings M R.Gene flow,mobile genetic elements and the recruitment of antibiotic resistance genes into Gram-negative pathogens[J].FEMS Microbiol Rev,2011,35(5):790-819.
[6]Neyra R C,Frisancho J A,Rinsky J L,et al.Multidrugresistant and methicillin-resistant Staphylococcus aureus(MRSA)in hog slaughter and processing plant workers and their community in North Carolina(USA)[J].Environ Health Persp,2014,122(5):471-477.
[7]Popowska M,Rzeczycka M,Miernik A,et al.Influence of soil use on prevalence of tetracycline,streptomycin,and erythromycin resistance and associated resistance genes[J].Antimicrob Agents Chemother,2012,56(3):1434-1443.
[8]季芳.基因芯片在细菌及其耐药检测中的应用[J].国际检验医学杂志,2011,32(2):240-242.
[9]Bruant G,Maynard C,Bekal S,et al.Development and validation of an oligonucleotide microarray for detection of multiple virulence and antimicrobial resistance genes in Escherichiacoli [J].Appl Environ Microbiol,2006,72(5):3780-3784.
[10]Handelsman J,Rondon M R,Brady S F,et al.Molecular biological access to the chemistry of unknown soil microbes:a new frontier for natural products[J].Chem Biol,1998,5(10):R245-R249.
[11]Costea P I,Zeller G,Sunagawa S,et al.Towards standards for human fecal sample processing in metagenomic studies [J].Nat Biotechno,2017,35:1069-1076.
[12]Schmieder R,Edwards R.Insights into antibiotic resistance through metagenomic approaches[J].Future Microbiol,2012,7(1):73-89.
[13]Chambers L,Yang Ying,Littier H,et al.Metagenomic analysis of antibiotic resistance genes in dairy cow feces following therapeutic administration ofthird generation cephalosporin[J].PLoSOne,2015,10(8):e0133764.
[14]Ma L,Xia Yu,Li Bing,et al.Metagenomic assembly reveals hosts of antibiotic resistance genes and the shared resistome in pig,chicken,and human feces[J].Environ Sci Technol,2015,50(1):420-427.
[15]Fang Hua,Wang Huifang,Cai Lin,et al.Prevalence of antibiotic resistance genes and bacterial pathogens in long-term manured greenhouse soils as revealed by metagenomic survey [J].Environ Sci Technol,2015,49(2):1095-1104.
[16]Stepanauskas R.Single cell genomics:an individual look at microbes[J].Curr Opin Microbiol,2012,15(5):613-620.
[17]王铱,徐鹏,戴欣.微生物单细胞基因组技术及其在环境微生物研究中的应用[J].微生物学报,2016,56(11):1691-1698.
[18]Nishikawa Y,Hosokawa M,Maruyama T,et al.Monodisperse picoliter droplets for low-bias and contamination-free reactions in single-cell whole genome amplification[J].PLoS One,2015,10(9):e0138733.
[19]Rinke C,Schwientek P,Sczyrba A,et al.Insights into the phylogeny and coding potential of microbial dark matter [J].Nature,2013,499(7459):431-437.
[20]祁彦洁,刘菲.地下水中抗生素污染检测分析研究进展[J].岩矿测试,2014,33(1):1-11.
[21]李圆杏,黄宏,刘臻.模拟日光照射下三种抗生素的光降解行为[J].环境化学,2013,32(8):1513-1517.
[22]沈怡雯,黄智婷,谢冰.抗生素及其抗性基因在环境中的污染、降解和去除研究进展[J].应用与环境生物学报,2015,21(2):181-187.
[23]张树清,张夫道,刘秀梅,等.高温堆肥对畜禽粪中抗生素降解和重金属钝化的作用[J].中国农业科学,2006,39(2):337-343.
[24]Wang L,Oda Y,Grewal S,et al.Persistence of resistance to erythromycin and tetracycline in swine manure during simulated composting and lagoon treatments[J].Microb Ecol,2012,63:32-40.
[25]Ho Y B,Zakaria M P,Latif P A,et al.Degradation of veterinary antibiotics and hormone during broiler manure composting [J].Bioresource Technol,2013,131:476-484.
[26]Arikan O,Mulbry W,Ingram D,et al.Minimally managed composting of beef manure at the pilot scale:effect of manure pile construction on pile temperature profiles and on the fate of oxytetracycline and chlortetracycline[J].BioresourceTechnol,2009,100(19):4447-4453.
[27]Engemann C A,Keen P L,Knapp C W,et al.Fate of tetracycline resistance genes in aquatic systems:Migration fromthe water column to peripheral bio films[J].Environ Sci Technol,2008,42:5131-5136.
[28]Forsberg K J,Patel S,Gibson M K,et al.Bacterial phylogeny structures soil resistomes across habitats[J].Nature,2014,59:612-616.
[29]Cheng Weixiao,Chen Hong,Su Chao,et al.Abundance and persistence of antibiotic resistance genes in livestock farms:A comprehensive investigation in eastern China[J].Environment International,2013,61(8):1-7.
[30]Wang Jian,Ben Weiwei,Zhang Yu,et al.Effects of thermophilic composting on oxytetracycline,sulfamethazine,and their corresponding resistance genes in swine manure[J].Environ Sci:Processes Impacts,2015,17(9):1654-1660.
[31]郑宁国,黄南,王卫卫,等.高温堆肥过程对猪粪来源抗生素抗性基因的影响[J].环境科学,2016,37(5):1986-1992.
猜你喜欢
军事文摘(2022年16期)2022-08-24
保健医苑(2022年5期)2022-06-10
现代临床医学(2022年3期)2022-06-06
昆明医科大学学报(2022年1期)2022-02-28
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
科学大众(2020年12期)2020-08-13
中国现代中药(2019年5期)2019-07-03
