
基质分散固相萃取-液相色谱-串联质谱法检测动物源性食品中硝基咪唑药物及其代谢物
2018-05-29 02:18:44邱凤梅余新威张志超
色谱 2018年5期
方 力, 邱凤梅, 余新威, 张志超
(1. 浙江省海产品健康危害因素关键技术研究重点实验室, 舟山市疾病预防控制中心, 浙江 舟山 316021; 2. 岱山县疾病预防控制中心, 浙江 岱山 316200)
硝基咪唑类药物是一类抗生素和抗原虫药,主要用于预防和治疗家禽组织滴虫病、球虫病以及猪出血性肠炎等疾病,其作为兽药广泛应用于畜禽和水产养殖业。硝基咪唑类药物主要包括甲硝唑(metronidazole, MNZ)、二甲硝基咪唑(dimetridazole, DMZ)、洛硝哒唑(ronidazole, RNZ)、奥硝唑(ornidazole, ONZ)、替硝唑(tinidazole, TNZ)、塞克硝唑(secnidazole, SNZ)和甲硝唑的代谢物羟基甲硝唑(hydroxyl metronidazole, MNZ-OH)以及二甲硝基咪唑和洛硝哒唑的共同代谢物1-甲基-5-硝基-2-羟甲基咪唑(2-hydroxymethyl-1-methyl-5-nitroimidazole, HMMNI)等。然而这些化合物被认为可能对人类产生致癌、致畸、致突变作用,已被美国和欧盟国家禁止使用[1,2]。我国农业部将二甲硝基咪唑和甲硝唑列为允许作治疗用、但不得在动物源性食品中检出的兽药[3]。因此,为了进一步加强相关食品安全问题的监管,开发简单、准确、快速检测动物源性食品中硝基咪唑类药物及其代谢物的方法显得十分必要。
目前,硝基咪唑类药物残留的检测方法主要有酶联免疫法[4-6]、气相色谱-质谱联用法[7,8]、毛细管电泳法[9]、高效液相色谱法[10,11]及液相色谱-串联质谱法[1,2,12-15]。酶联免疫法易产生假阳性或者假阴性结果,一般用于常规大批量快速筛查;气相色谱-质谱联用法需要衍生化,实验条件要求严格;毛细管电泳法、液相色谱法灵敏度相对较低;液相色谱-串联质谱法(HPLC-MS/MS)具有选择性好、灵敏度高等优势。当前关于食品中硝基咪唑类药物残留检测的前处理手段主要有固相萃取净化法、分子印迹净化法等[1,2,12-15]。常规固相萃取净化法存在溶剂消耗量大、使用成本高等缺陷;分子印迹净化法尽管净化效果好,但也存在操作要求高、制备时间长等缺陷。近年来,分散固相萃取技术(dispersive solid phase extraction, dSPE)广泛应用于多种类样品的前处理,该净化手段具有快速、高效、操作简单、成本低等特点,可实现高通量检测[16-18]。
本研究采用基质分散固相萃取技术结合HPLC-MS/MS,建立了测定鸡蛋、鸡肉、鱼肉和猪肉等动物源性食品中8种硝基咪唑类药物及其代谢物残留的方法。
1 实验部分
1.1 仪器、试剂与材料
TSQ Vantage三重四极杆质谱仪(配有电喷雾离子(ESI)源)、Ultimate 3000超高效液相色谱仪(美国Thermo公司); Allegra X-15R型高速冷冻离心机、Microfuge 20型微量高速离心机(美国Beckman公司); DMT-2500型多管涡旋混合器(杭州米欧仪器公司); GM200型刀式研磨仪(德国Retsch公司); Milli-Q(18.2 MΩ5cm)超纯水处理系统(德国Merck公司)。
标准品:MNZ、DMZ、HMMNI、RNZ、ONZ、TNZ、氘代甲硝唑(MNZ-D4)、氘代替硝唑(TNZ-D5)均购自德国Dr. Ehrenstorfer公司;SNZ购自日本TCI公司;MNZ-OH购自德国Witega公司,标准品纯度均≥98%;甲酸(LC-MS级)、乙酸乙酯(色谱纯)和乙腈(色谱纯)均购自德国CNW公司。
乙二胺基-N-丙基硅烷填料CNWBOND PSA(40~63 μm)购自德国CNW公司;孔径0.22 μm亲水聚四氟乙烯(PTFE)滤膜购自上海安谱有限公司。超纯水由Milli-Q超纯水系统制得。
1.2 样品前处理
1.2.1样品采集
鸡肉、鸡蛋、猪肉、罗非鱼肉均购自当地农贸市场或超市,取500 g基质样品经刀式研磨仪均质后,于-20 ℃冷冻保存,待用。
1.2.2提取
准确称取(2.00±0.01) g均质后样品,置于50 mL聚丙烯离心管中,加入10 mL乙酸乙酯,涡旋混合10 min,超声提取10 min,以8 000 r/min离心5 min,移取上清液至另一25 mL离心管中,再向残渣中加入10 mL乙酸乙酯,重复以上提取步骤,合并提取液,于40 ℃水浴下氮吹至近干,用2 mL 0.1%(体积分数)甲酸水溶液溶解,待净化。
1.2.3净化
向提取液中加入4 mL正己烷,涡旋混匀1 min,以8 000 r/min离心5 min,弃去正己烷层,移取1.0 mL提取液,置于已装有(50±3) mg PSA粉末的离心管中,涡旋混匀1 min,以15 000 r/min离心5 min,上清液经0.22 μm滤膜过滤,注入样品瓶中,备用。
称取不含目标化合物的基质样品于离心管中,按照上述步骤进行提取和净化,得到空白基质溶液。
1.3 标准溶液的配制
准确称取8种硝基咪唑类药物及其代谢物标准品,用乙腈溶解,配制成100 mg/L的标准储备液。分别移取适量标准储备液,用0.1%(体积分数)甲酸水溶液稀释,配制成1.0 mg/L混合标准中间液;最后用空白基质溶液配制成质量浓度为2、5、10、20和40 μg/L的系列混合标准工作液。
1.4 分析条件
色谱柱:Hypersil GOLD C18柱(100 mm×2.1 mm, 1.9 μm,美国Thermo公司);柱温:30 ℃;流动相:A为0.1%(体积分数)甲酸水溶液,B为乙腈;流速:0.3 mL/min。梯度洗脱程序:0~2.0 min, 8%B; 2.0~7.0 min, 8%B~70%B; 7.0~7.2 min, 70%B~8%B; 7.2~10.0 min, 8%B。进样量:10 μL。
离子源:电喷雾离子(ESI)源,正离子模式;扫描模式:选择反应监测(SRM)模式;离子传输温度:325 ℃;喷雾电压:3.0 kV;汽化温度:300 ℃;鞘气(氮气)压力:45 arb;辅助气(氮气)压力:15 arb;碰撞气:高纯氩气,压力0.2 Pa。8种硝基咪唑类药物及其代谢物的其他质谱参数见表1。

表 1 硝基咪唑类药物及其代谢物的质谱参数
*Quantitative ion.
2 结果与讨论
2.1 分析条件的优化
2.1.1色谱条件的优化
硝基咪唑类药物及其代谢物是一种中等极性化合物,已有文献[1,2]证实反相色谱柱更适合该类化合物的分离与检测。本实验比较了Hypersil GOLD C18(100 mm×2.0 mm, 1.9 μm)和Hypersil GOLD aQ(100 mm×2.0 mm, 1.9 μm)反相色谱柱对目标化合物的分离效果。结果显示,与Hypersil GOLD aQ色谱柱相比,Hypersil GOLD C18色谱柱对目标化合物的分离效果更好,色谱峰峰形更尖锐。
本研究还比较了0.1%(体积分数)甲酸水-乙腈与含5 mmol/L甲酸铵的0.1%(体积分数)甲酸水-乙腈两个流动相体系对目标化合物分离效果的影响。结果显示,当流动相体系中加入甲酸铵后,部分目标物的色谱峰出现峰展宽的现象,响应强度下降。综上所述,本研究采用Hypersil GOLD C18色谱柱,0.1%(体积分数)甲酸水-乙腈作为流动相分离待测组分,此时目标化合物能较好地实现分离,其色谱图见图1。
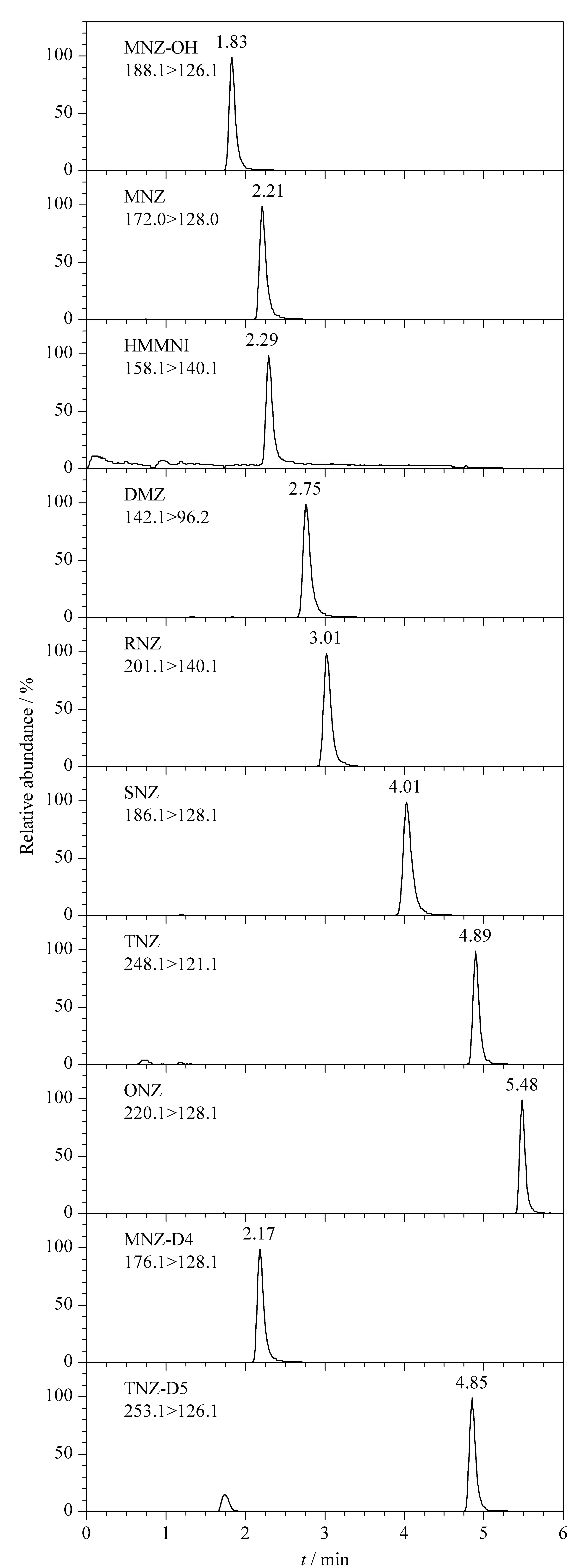
图 1 在SRM模式下硝基咪唑类药物及其代谢物的色谱图Fig. 1 Chromatograms of the nitroimidazoles and their metabolites in the selected reaction monitoring (SRM) mode
2.1.2质谱条件的优化
实验中以1.0 mg/L混合标准中间液为对象,采用流动注射的方式在ESI+模式下进行质谱参数的优化。选择干扰小、信噪比大的离子对作为定性或定量离子对。优化后的透镜电压、碰撞能量、定性和定量离子对见表1。选出的离子对数量符合欧盟EC/657指令中利用质谱方法对药物残留进行确证时必须满足4个识别点的要求(母离子为1个识别位点,特征子离子为1.5个识别位点)。
2.2 前处理条件的优化
2.2.1提取条件的优化
硝基咪唑类化合物的提取试剂一般为乙腈、乙酸乙酯、二氯甲烷和甲醇等,本文对比了分别采用甲醇、乙腈、二氯甲烷、乙酸乙酯、二氯甲烷-乙腈(1∶1, v/v)时目标化合物的提取效率(见图2)。可以看出,提取溶剂对MNZ-OH提取回收率的影响最为明显,采用乙酸乙酯提取时,回收率最高,为90.7%;提取溶剂对其他目标化合物的影响不显著。实际样品存在大量水溶性化合物,乙腈、甲醇等溶剂易溶于水,容易将水溶性杂质带入提取液中;采用乙酸乙酯萃取,大部分亲水性杂质被去除,可降低后续净化的难度。因此,本研究选用乙酸乙酯作为提取溶剂。

图 2 提取溶剂对硝基咪唑类药物及其代谢物回收率的影响(n=3)Fig. 2 Effects of extraction solvents on the recoveries of the nitroimidazoles and their metabolites (n=3)
2.2.2净化条件的优化
动物源性食品中的脂肪含量较高,样品经有机溶剂提取后,大部分脂肪被提取出来。对样品提取液中脂肪的去除是样品前处理过程中非常重要的步骤。本研究考察了正己烷、石油醚、环己烷3种溶剂对脱脂净化效果的影响。结果显示,3种脱脂溶剂都能达到较好的净化效果,鸡蛋基质样品的变化尤其明显。鸡蛋基质中含有大量脂溶性色素,样品提取液呈黄色,经脱脂溶剂净化后,样品溶液变为无色透明。综合考虑脱脂溶剂的毒性和使用成本,本研究采用最常用的正己烷作为脱脂溶剂。
基质分散固相萃取是一种简单高效的净化方法,其原理是利用固体吸附剂选择性吸附杂质从而达到净化样品的目的。吸附剂的选择是dSPE的一个最重要参数。C18填料是一种憎水硅胶基吸附剂,其对非极性化合物具有很强的吸附性能;HLB填料是一种以改性的二乙烯基苯聚合物为基质的吸附剂,其亲水-亲脂平衡性能的设计适用于酸性、碱性和中性化合物的净化;石墨化炭黑(GCB)具有六元环结构,因此与平面分子具有很强的亲和力,适用于多种有机化合物的提取和净化,特别适用于从各种基质中分离和除去有色物质和固醇类等。PSA填料的主要成分是乙二胺-N-丙基硅烷,能与样品中有机酸、色素、金属离子、酚类化合物结合以去除基质中的相关干扰物。本研究考察了CNWBOND HC-C18(40~63 μm)、Poly-Sery HLB(20~120 μm)、CNWBOND Carbon-GCB(37~125 μm)和CNWBOND PSA(40~63 μm)吸附剂对硝基咪唑类药物及其代谢物的吸附情况。如图3所示,GCB和HLB填料对目标化合物的吸附作用较强,目标化合物的回收率小于50%; C18填料对目标物也产生了一定的吸附作用,且离心后部分C18填料仍处于悬浮状态,对后续取样造成了困难;采用PSA吸附剂时目标物的回收率最高。因此实验最终选用PSA吸附剂。
实验还对比了不同PSA吸附剂用量(25、50、100和200 mg)对目标化合物回收率的影响。结果发现,除采用25 mg PSA吸附剂产生的净化效果较差外,不同PSA吸附剂用量对硝基咪唑类药物及其代谢物回收率的影响并不明显。因此最终确定PSA吸附剂用量为50 mg。
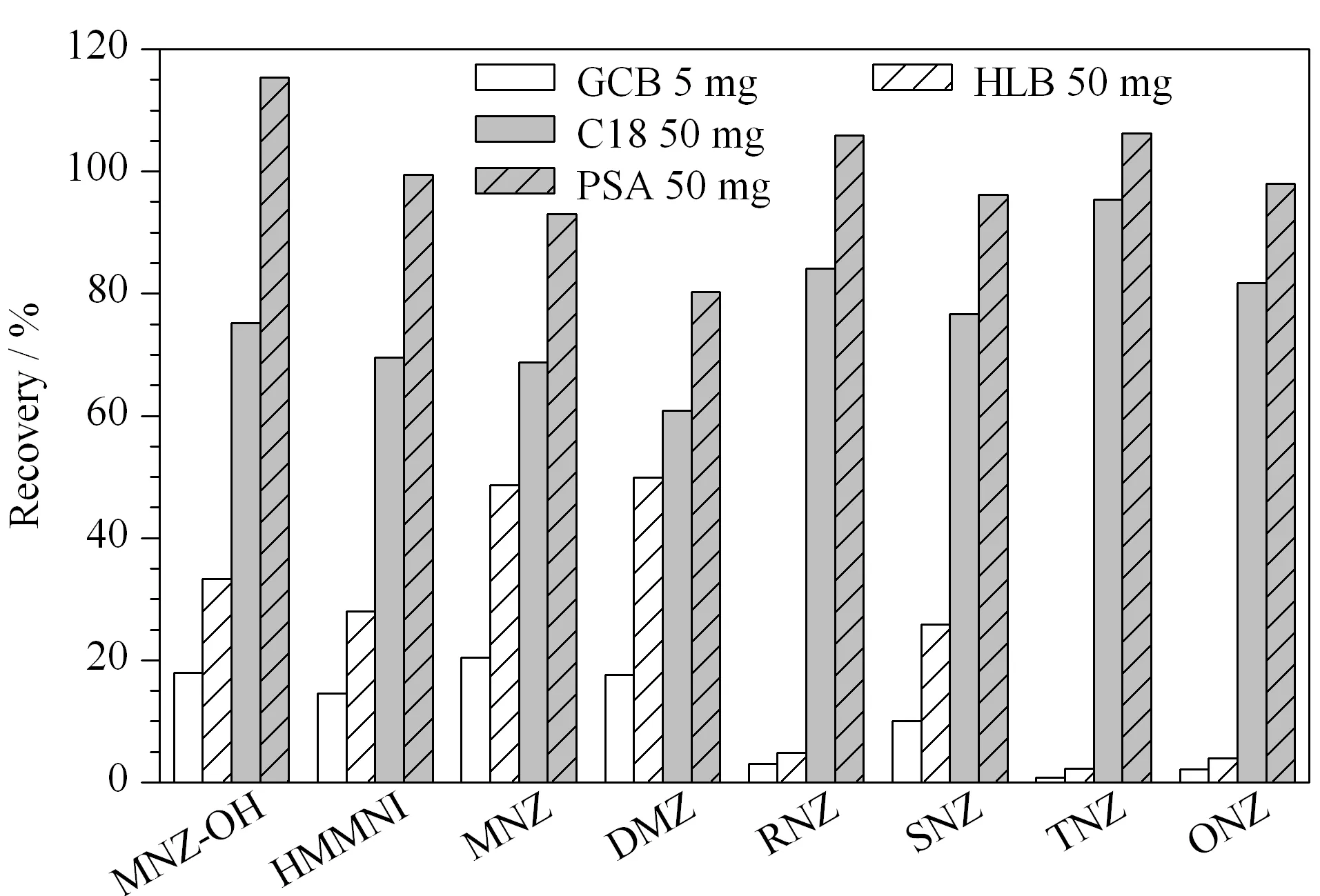
图 3 吸附剂对硝基咪唑类药物及其代谢物回收率的影响Fig. 3 Effects of adsorbents on the recoveries of the nitroimidazoles and their metabolites GCB: graphitized carbon black; HLB: hydrophilic-lipophilic balance; PSA: primary-secondary amine.
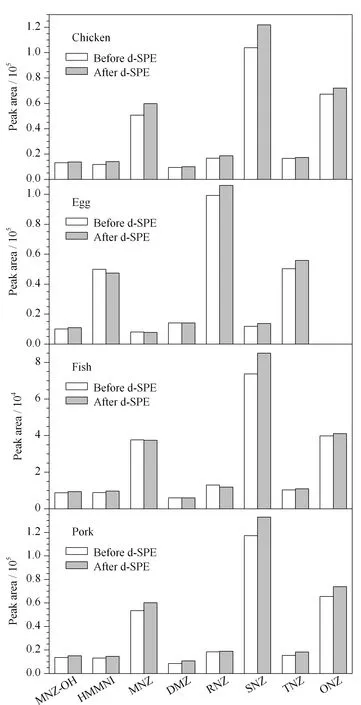
图 4 分散基质固相萃取前后目标化合物的峰面积Fig. 4 Peak areas of the target compounds before and after dispersive solid phase extraction (dSPE)
为了证明PSA吸附剂的净化效果,本研究比较了在净化前后4种不同动物源性食品基质加标样品(5 μg/kg)中各硝基咪唑类药物及其代谢物的峰面积。由图4可知,经PSA吸附剂净化后,大多数目标化合物的绝对响应峰面积有所增加,猪肉基质中DMZ的响应值提高最明显,达25.9%; 4种基质中HMMNI和SNZ的响应也均有10%以上的提高。
2.3 基质效应(ME)评估
本研究按(空白基质匹配标准曲线斜率/纯溶剂标准曲线斜率-1)×100%的计算方法对基质效应进行评价(见表2)。当ME为负值时表示存在基质抑制效应,ME为正值时表示存在基质增强效应;绝对值越大则基质效应越强,如恰好为0则表示不存在基质效应。由表2可知,硝基咪唑类药物及其代谢物在各基质溶液中呈现一定的基质增强作用,尤其是猪肉样品基质增强效应最为明显,最高为+22.5%,其他样品基质中各硝基咪唑类药物及其代谢物的基质效应为-10.9%~+16.8%。对于复杂的食品类基质而言,经本方法净化处理后目标化合物的基质效应均在可接受范围内。本文采用基质匹配内标法测定实际样品也可在一定程度上补偿基质效应的影响。
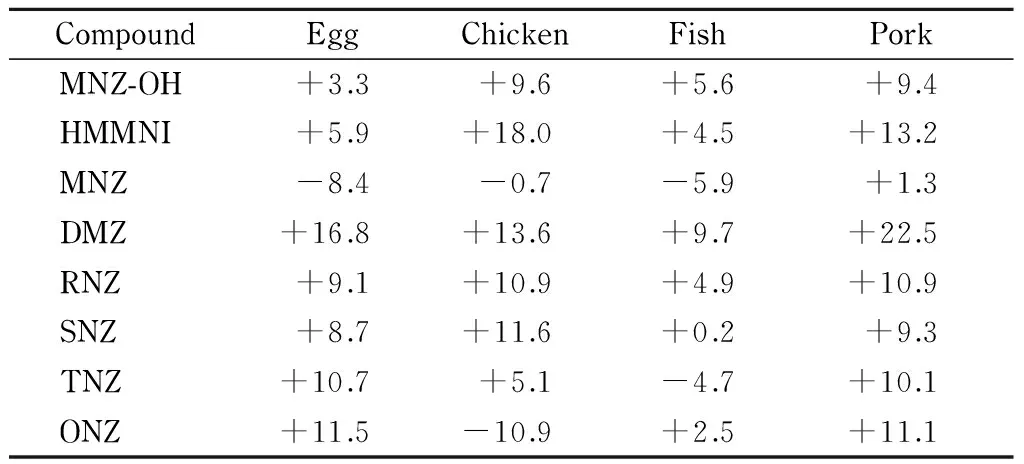
表 2 不同基质中硝基咪唑类药物及其代谢物的基质效应
2.4 标准曲线和检出限
按1.3节方法配制基质标准溶液,并对其进行分析,以硝基咪唑类药物及其代谢物的质量浓度为横坐标、目标化合物峰面积与内标峰面积的比值为纵坐标绘制基质工作曲线(见表3)。结果表明,在0.5~20 μg/L范围内目标化合物线性相关性良好,相关系数(r2)均≥0.998 2。
在本实验条件下,根据3倍信噪比下目标化合物色谱峰的响应值,得出4种动物源性食品基质中MNZ-OH和HMMNI的检出限分别为0.2 μg/kg和0.5 μg/kg,其他目标化合物的检出限为0.1 μg/kg; MNZ-OH和HMMNI的定量限分别为0.5 μg/kg和1.0 μg/kg,其他目标化合物的定量限为0.3 μg/kg。
2.5 回收率与精密度
准确称取空白鸡蛋、鸡肉、鱼肉和猪肉样品各2.0 g,设定3个添加水平(1.0、3.0和10.0 μg/kg),根据1.2节和1.3节方法进行检测,每组6个平行样品,测定回收率和相对标准偏差。由表4可知,各基质中硝基咪唑类药物及其代谢物的回收率为84.2%~120.8%,相对标准偏差为2.0%~16.2%(n=6)。
2.6 实际样品检测
应用本方法对2016~2017年舟山地区采集的15份鸡蛋和15份鸡肉样品进行检测,其中有1份鸡蛋样品检出甲硝唑和羟基甲硝唑,含量分别为3.49 μg/kg和2.43 μg/kg,其余样品均未检出硝基咪唑类药物及其代谢物。

表 4 不同基质中硝基咪唑类药物及其代谢物的回收率和精密度(n=6)

CompoundSpiked/(μg/kg)EggRecovery/%RSD/%ChickenRecovery/%RSD/%FishRecovery/%RSD/%PorkRecovery/%RSD/%MNZ-OH1.0110.612.495.28.290.37.797.09.43.0101.67.390.26.589.23.788.26.310.098.65.589.94.193.32.991.03.3HMMNI1.0100.013.8103.27.098.716.295.99.63.0100.29.7100.66.197.25.395.58.610.0103.66.7109.33.9103.75.199.17.1MNZ1.0120.82.0112.14.5110.42.4103.210.73.0112.97.6107.25.0107.62.6102.83.810.0107.55.4108.43.4107.82.8100.44.5DMZ1.096.214.190.97.9107.02.484.212.13.096.610.687.67.9103.83.789.15.410.095.34.291.96.8106.84.688.66.2RNZ1.096.412.5103.75.684.39.887.012.23.099.16.3103.23.0106.93.495.24.610.0106.98.9105.04.7104.64.393.73.0SNZ1.0104.87.4102.93.7109.99.4102.69.03.0103.46.6100.15.2106.75.697.59.610.0102.13.5103.33.4111.44.495.65.0TNZ1.0103.010.091.615.299.212.0105.314.53.0101.66.692.05.3106.22.994.810.410.0103.15.6108.04.6107.55.886.05.3ONZ1.088.45.699.84.7111.45.4104.911.93.089.13.896.92.7108.62.294.15.410.091.16.296.55.3113.13.089.44.3
3 结论
本方法采用乙酸乙酯提取,正己烷脱脂,基质分散固相萃取净化,液相色谱-串联质谱检测,建立了准确定性定量检测动物源性食品中8种硝基咪唑类药物及其代谢物残留的方法。本方法成本低,操作简便,灵敏度高,重复性好,能够用于应急检测或日常兽药残留监测。
:
[1] Cronly M, Behan P, Foley B, et al. J Chromatogr A, 2009, 1216(46): 8101
[2] Xia X, Li X W, Zhang S X, et al. J Chromatogr A, 2008, 1208(1/2): 101
[3] Ministry of Agriculture. No. 235 Bulletin of the Ministry of Agriculture of the People’s Republic of China. (2002-12-24) [2017-12-20]. http://www.moa.gov.cn/zwllm/nybz/200803/t20080304_1028649.htm
农业部. 中华人民共和国农业部公告第235号. (2002-12-24) [2017-12-20]. http://www.moa.gov.cn/zwllm/nybz/200803/t20080304_1028649.htm
[4] Zhang C, Pan J R, Shuai R Q, et al. Journal of Nuclear Agricultural Sciences, 2016, 30(2): 323
张弛, 潘家荣, 帅瑞琪, 等. 核农学报, 2016, 30(2): 323
[5] Wang Y B, He F Y, Wan Y P, et al. Food Addit Contam A, 2011, 28(5): 619
[6] Thompson C S, Traynor I M, Fodey T L, et al. Anal Chim Acta, 2009, 637(1/2): 259
[7] Wang J C, Ma S Y, Wang D J, et al. Chinese Journal of Animal and Veterinary Sciences, 2008, 39(12): 1772
汪纪仓, 马素英, 王大菊, 等. 畜牧兽医学报, 2008, 39(12): 1772
[8] He D, Li X Y, Xian Y P, et al. Journal of Instrumental Analysis, 2015, 34(8): 911
何东, 李秀英, 冼燕萍, 等. 分析测试学报, 2015, 34(8): 911
[9] Hernandez-Mesa M, Garcia-Campana A M, Cruces-Blanco C. Food Chem, 2014, 145(4): 161
[10] Wang Y, Zheng C Y, He F, et al. Food Science, 2011, 32(20): 197
王杨, 郑重莺, 何丰, 等. 食品科学, 2011, 32(20): 197
[11] Lu K, Tong Q Y. Journal of Instrumental Analysis, 2011, 30(11): 1320
卢坤, 童群义. 分析测试学报, 2011, 30(11): 1320
[12] Li C Y, Wu M, Yan L J, et al. Journal of Food Safety and Quality, 2012, 3(1): 17
黎翠玉, 吴敏, 严丽娟, 等. 食品安全质量检测学报, 2012, 3(1): 17
[13] Guo X C, Xia Z Y, Wang H H, et al. Talanta, 2017, 166: 101
[14] Zhang L, Kong X H, Wang H, et al. Chinese Journal of Analytical Chemistry, 2014, 42(12): 1735
张璐, 孔祥虹, 王菡, 等. 分析化学, 2014, 42(12): 1735
[15] Liu Y M, Gao Y Z, Li J, et al. Chinese Journal of Chromatography, 2010, 28(6): 596
刘永明, 曹彦忠, 李金, 等. 色谱, 2010, 28(6): 596
[16] Zhang Y, Yue Z F, Lan F, et al. Chinese Journal of Analytical Chemistry, 2012, 40(5): 724
张毅, 岳振峰, 蓝芳, 等. 分析化学, 2012, 40(5): 724
[17] Cui C Y, Zhang H Y, Wu X Q, et al. Chinese Journal of Chromatography, 2017, 35(5): 487
崔春艳, 张红医, 吴兴强, 等. 色谱, 2017, 35(5): 487
[18] Luo H T, Huang X L, Wu H Q, et al. Chinese Journal of Chromatography, 2017, 35(8): 816
罗辉泰, 黄晓兰, 吴慧勤, 等. 色谱, 2017, 35(8): 816
猜你喜欢
现代临床医学(2022年4期)2022-09-29 07:36:10
中学生学习报(2021年20期)2021-11-24 15:30:52
鞍钢技术(2021年3期)2021-06-11 05:13:50
化工管理(2021年7期)2021-05-13 00:46:24
中国农资(2016年1期)2016-12-01 05:21:23
分析测试学报(2015年8期)2016-01-13 06:19:31
分析测试学报(2015年7期)2016-01-13 06:19:16
化工进展(2015年3期)2015-11-11 09:08:25
质谱学报(2015年5期)2015-03-01 03:18:37
四川师范大学学报(自然科学版)(2015年3期)2015-02-28 14:08:00
