
IgG4相关性疾病累及胰腺、胆道、肝脏和淋巴结1例病例报告
2018-05-10 22:49:44崔晓丙吴保平
现代消化及介入诊疗 2018年1期
崔晓丙 冯 洁 吴保平
IgG4相关性疾病(immunoglobulin G4 associated disease,IgG4-RD)是一种可以累计全身多个器官的慢性炎性伴纤维化的疾病,临床表现为血清IgG4水平升高和受累组织中弥漫性或局灶性肿大、硬化,病理特点为受累组织中以IgG4阳性浆细胞为主的淋巴细胞浸润、闭塞性静脉炎和席纹状纤维化。IgG4-RD致病机制尚不明确,目前考虑其发病可能与遗传易感性,环境因素和细菌感染等诱导的过敏反应或免疫异常相关[1]。
IgG4相关性疾病通常累及多处器官,临床特征表现特异,容易误诊漏诊。南方医院消化内科于2017年9月收治1例IgG4相关性疾病患者,主要累及胰腺、胆道、肝脏和淋巴结,现报道如下。
一、病历资料
患者,男性,52岁,因“皮肤、巩膜黄染,体重下降2月余”于2017-09-04入院。
患者于2017年6月无明显诱因出现皮肤、巩膜黄染,尿黄,无腹痛。遂至外院(A)就诊,彩超提示肝内回声较密、较粗;胰管增宽;胰头部低回声性质待定;胆总胆管上段及肝内胆管扩张。MRI提示自身免疫性胰腺炎并胆系扩张。增强CT提示自身免疫性胰腺炎合并硬化性胆管炎。免疫球蛋白IgG4 13.6 g/L(↑)。诊断为“自身免疫性胰腺炎合并硬化性胆管炎”,对症治疗后好转出院,未予激素治疗。
2017年7月患者无明显诱因出现腹痛、发热,外院(B)就诊,彩超检查提示腹腔积液,考虑“消化道穿孔”,行手术治疗后好转出院,手术过程不详。出院后复查CT检查提示肝左外叶低密度占位,腹腔内及腹膜后多发淋巴结肿大,双侧胸腔少量积液,考虑“肝脓肿”可能,治疗不详。
2017年8月患者再次出现皮肤、巩膜黄染、尿黄,外院(B)增强CT示:肝内S2段低密度肿物,考虑为原发性肝癌可能;肝总管下段鼠尾状狭窄及以上肝内、外轻度扩张且轻度强化,考虑慢性胆管炎改变;少量腹水;少量胸水。 诊断为“原发性肝癌”,未予特殊治疗(患者拒绝手术)。
2017-09-04患者就诊于我院肝胆外科,外科拟“原发性肝癌”肝功能异常不宜手术转入我科。自发病以来,患者精神差、纳减、睡眠差、大便正常、体重下降约20 kg。患者既往糖尿病史数年(未治疗),不明药物过敏史,吸烟指数600(已戒烟2月),无急性胰腺炎、高脂血症、乙肝病史,无暴饮暴食及长期饮酒史。入院查体:身目黄染,腹部可见纵行手术瘢痕,无压痛,肝脾肋下未及。
血清学检查 (异常结果):WBC 11.5×109/L,EOS 4.81×109/L,EOS 41.8% ,ALT 99 U/L,AST 124 U/L,TBIL 74.7 μmol/L,DBIL 35 μmol/L,ALP 719 U/L,γ-GT 408 U/L,TBA 14.3 μmol/L,CA-199 161.78 U/mL,血清 IgG4 21.1 g/L,尿胆红素(2+),尿胆原(2+)。
腹部超声提示:肝脏增大(右叶斜径15.5 cm,肋下2.4 cm),实质回声增粗,分布欠均匀;肝S2段6.1×2.8 cm片状低回声区,边缘模糊,网格状改变;肝内胆管扩张,胆总管上段扩张(1.3 cm);腹膜后实性占位低回声团,与胰头界限不清;腹腔少量积液。超声诊断考虑胆管细胞CA与肝脓肿相鉴别,腹膜后肿大淋巴结。超声造影(声诺维3 mL)提示:肝S2段7.4×4.0 cm片状低回声区,动脉期等增强,动脉晚期早消退,门脉期及延迟期呈低增强,病变其中肝内胆管扩张(1.4 cm)。超声造影诊断考虑胆管细胞CA伴胆管扩张。行超声引导下肝占位经皮穿刺活检。肝占位病理示:肝细胞水肿,胆汁淤积,免疫组化示汇管区较多IgG4阳性浆细胞浸润,IgG4与IgG阳性细胞比值超过40%,IgG4阳性浆细胞>10个/HPF,考虑为IgG4相关硬化性胆管炎(图1)。
MRI增强(普美显)提示:肝左叶欠规整结节影,长T1长T2信号;GD-EOB-DTPA增强见外周环形强化,门脉期及延迟期外周环形强化;DWI相稍高信号,ADC图低信号;腹腔、腹膜后多发肿大淋巴结(大者长径1.2 cm);腹水;胰腺无明显异常。MRI增强诊断考虑胆管细胞CA与脓肿相鉴别。MRCP提示:肝内外胆管扩张、增粗,管壁增厚,胆总管下段呈鼠尾样改变。MRCP诊断考虑硬化性胆管炎合并感染(图2)。
ERCP提示:十二指肠乳头明显肿胀,左肝内胆管及胆总管上段扩张,边缘光滑,胆总管下段呈鼠尾状狭窄;胰管未见明显狭窄、扩张及结石影。ERCP术后留置胆道支架,并于胆总管下段乳头切开处取活检。ERCP诊断考虑胆总管下段炎性狭窄可能。乳头切开处活检病理提示:间质纤维组织增生,散在大量淋巴细胞、浆细胞、嗜酸性粒细胞及少许中性粒细胞浸润,免疫组化示IgG4与IgG阳性细胞比值超过40%,IgG4阳性浆细胞>10个/HPF,考虑为IgG4相关疾病 (图3)。
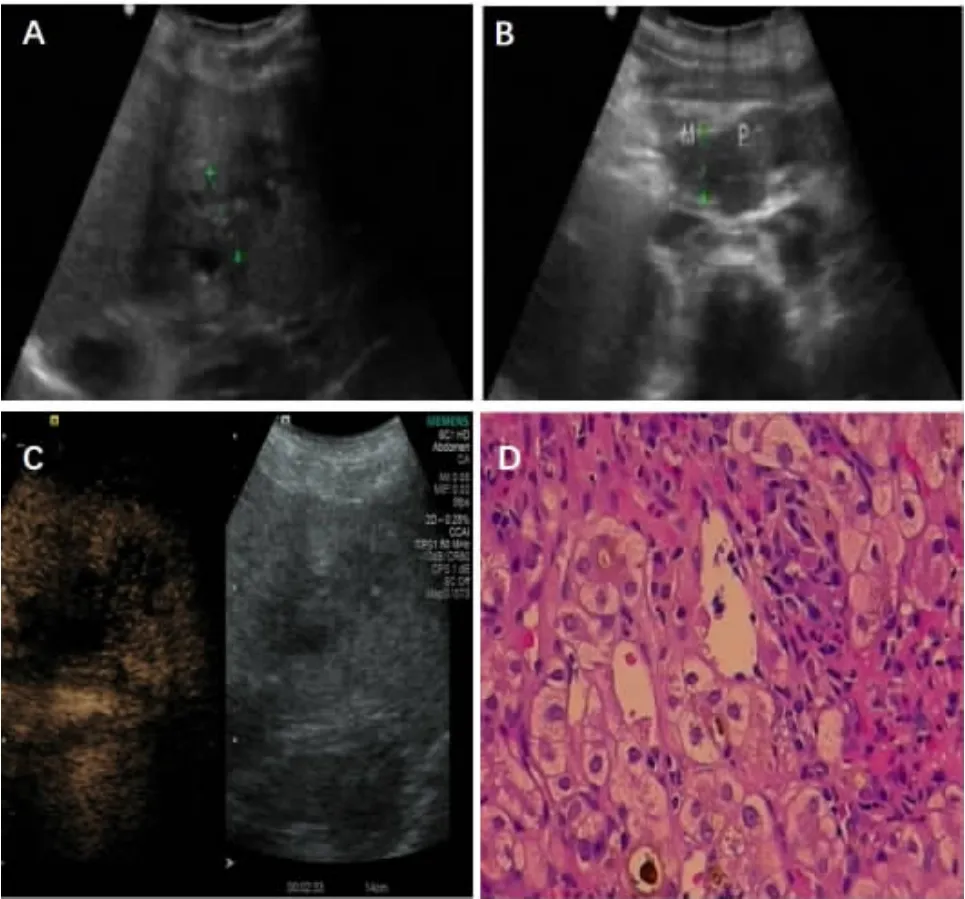
图1 腹部超声、超声造影及肝占位穿刺病理[(A)肝S2段6.1×2.8 cm片状低回声区;(B)腹膜后实性占位低回声团,与胰头界限不清;(C)超声造影示延迟期(153s)低增强;(D)穿刺病理提示肝细胞水肿,胆汁淤积,浆细胞浸润]
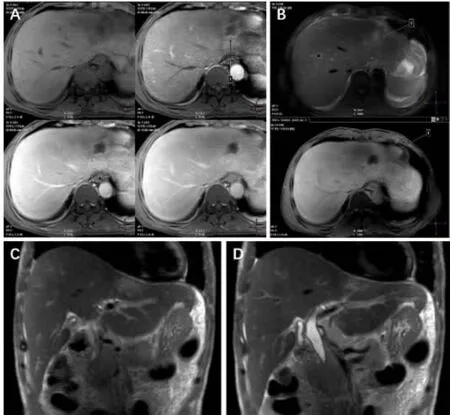
图2 MRI增强扫描及MRCP[(A)GD-EOB-DTPA增强见病变环周增强;(B)DWI相稍高信号,ADC 图低信号;(C、D)肝内外胆管扩张、增粗,管壁增厚,胆总管下段呈鼠尾样改变]
二、诊断、治疗及随访
结合以上资料,患者诊断为“①IgG4相关性疾病:IgG4硬化性胆管炎,IgG4相关肝损伤,IgG4相关肝内炎性假瘤,IgG4相关胰腺炎 (自身免疫性胰腺炎)? IgG4相关腹腔及腹膜后淋巴结肿大?②胆道感染。③腹水。④2型糖尿病。⑤中度嗜酸粒细胞血症。 ⑥剖腹探查术后”。在留置胆管支架基础上,予“泼尼松(30 mg QD,0.5 mg/Kg)+熊去氧胆酸(250 mg TID)”治疗,辅助抗生素、抑酸、促进消化动力、补充胰酶及降糖等对症治疗。
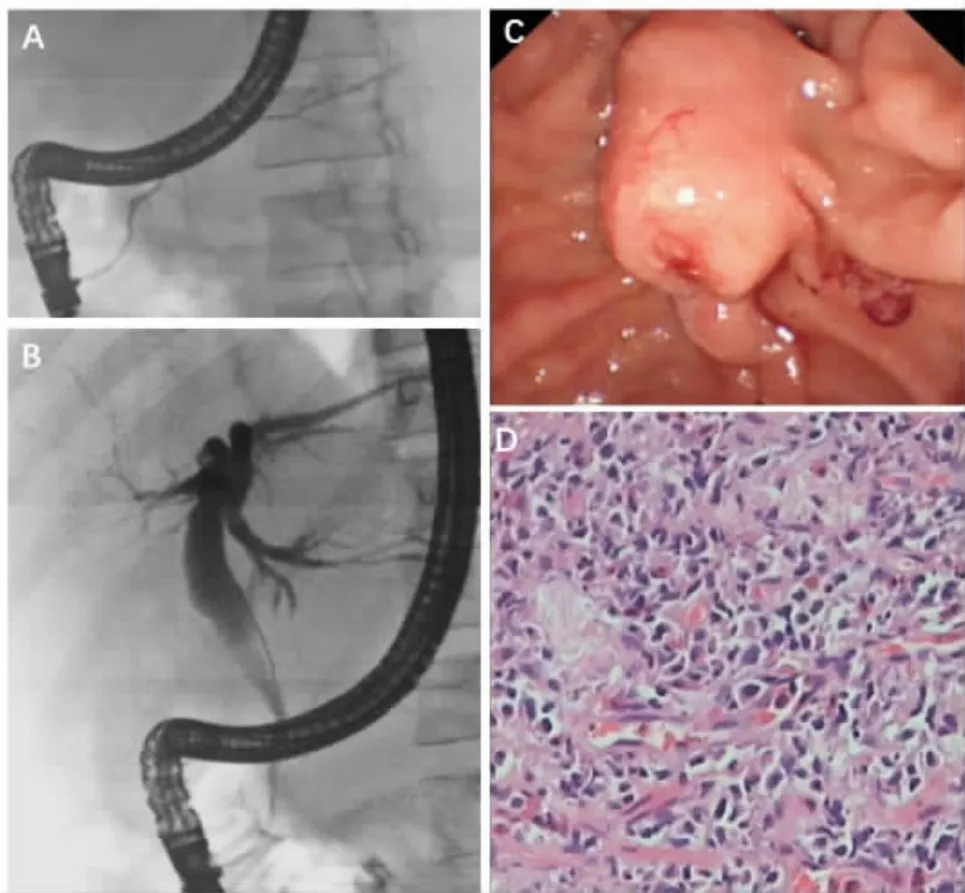
图3 ERCP及乳头切开处活检病理[(A)胰腺主、分支胰管走行正常,无明显狭窄、扩张及中断表现;(B)左肝内胆管及胆总管上段扩张,边缘光滑,胆总管下段呈鼠尾状狭窄;(C)十二指肠乳头明显肿胀;(D)纤维组织增生及大量浆细胞浸润]
服药 4周后复查:ALT、AST、TBIL、DBIL、CA-199、ALP、尿胆红素、尿胆原降至正常,γ-GT 95U/L、TBA 14 μmol/L、血清IgG4 11.7 g/L。腹部超声提示肝左外叶见一3.7×2.6 cm片状稍低回声区,肝内胆管未见明显扩张,胆总管上段扩张。增强CT提示肝左叶异常密度影范围基本同前,肝左叶肝内胆管及胆总管上段扩张、积气,原腹腔及腹膜后多发淋巴结较前减小。考虑肝占位范围无明显缓解,除抗生素外,继续维持原激素剂量治疗。
服药 8周后复查:ALT、AST、TBIL、DBIL、CA-199、ALP、γ-GT、TBA、尿胆红素、尿胆原正常,血清 IgG4 12.5 g/L。腹部超声提示肝左外叶2.8×1.8 cm片状稍低回声区,肝左外叶胆管扩张,胆总管上段增宽,原腹膜后肿大淋巴结未见。超声造影提示肝S2段3.2×2.4 cm片状低回声区,动脉晚期早消退,门脉期及延迟期呈低增强。再次超声引导下肝占位穿刺活检,病理提示肝组织结构尚规则,部分肝细胞水肿、体积较大,汇管区见少量淋巴细胞及少许浆细胞浸润;免疫组化示IgG4+/IgG+细胞大于40%(图4)。考虑临床症状、血清学好转,肝脏占位缓解,激素减量至 “泼尼松 (25 mg QD)”,辅助抑酸及补充胰酶治疗。
后续计划:激素继续减量(-5 mg Q2W)至5 mg QD维持治疗,定期复查肝功能及腹部超声,择期取出胆道支架。
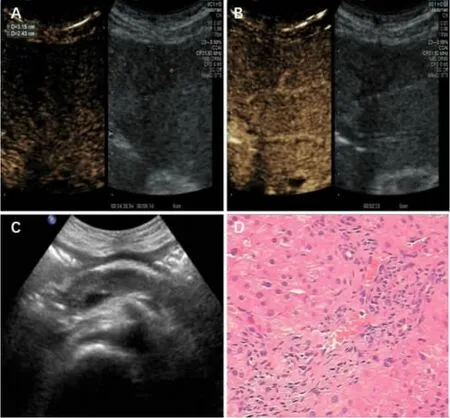
图4 激素治疗8周后复查超声及肝占位穿刺病理 [(A、B)肝S2段片状低回声区范围较前明显缩小,动脉晚期早消退,延迟期呈低增强;(C)胰腺回声减低,微小钙化灶形成;(D)肝细胞水肿较前明显好转,汇管区淋巴细胞及浆细胞浸润减少]
三、IgG-RD在胰腺、胆道和肝脏的表现和诊断标准
根据协和医院2017年报道,我国IgG4-RD常累及的器官有淋巴结、颌下腺、泪腺、胰腺、肺、胆道、鼻窦、腮腺、腹膜后、大动脉、肾脏、皮肤,少见受累包括甲状腺、垂体、胃肠道、神经系统、心包、纵隔和睾丸等。多数IgG4-RD患者为多器官受累,超过70%患者受累器官≥3个,超过20%患者2个器官受累[2]。
1.自身免疫性胰腺炎
自身免疫性胰腺炎(autoimmune pancreatitis,AIP)由Yoshida等在1995年正式命名,2001年Hamano等发现AIP与IgG4升高相关,随后2003年Kamisaw等报道胰腺外组织也存在IgG4+浆细胞浸润,提出了AIP是IgG4-RD的胰腺表现这一概念[3]。
基于组织病理学(histology)、影像学(imaging)、血清学(serology)、多器官累及(other organ involvement)和类固醇激素治疗有效(response to therapy)等五项,美国在2006年提出了AIP的HISORt诊断标准,并将其分为三个亚组,其中任意一组均可单独诊断AIP(表1)[4]。2011年国际共识诊断标准(International Consensus Diagnostic Criteria,ICDC)将 AIP 分为两个亚型:1型AIP为淋巴浆细胞硬化性胰腺炎(lymphoplasmacytic sclerosing pancreatitis,LPSP),亦称为 IgG4 相关胰腺炎;2型AIP型为特发性导管中心性胰腺炎(idiopathic duct-centric pancreatitis,IDCP)[5]。我国的共识意见参考了该标准,需要指出的是,因为2型AIP少见,文献报道中AIP一般指代1型AIP,本文亦采用类似表达[6]。
AIP常于老年男性发生,诊断时年龄大多处于50~60岁,男女性别比>3:1。临床症状以黄疸最常见(60%~75%),还有腹部隐痛不适、消瘦、腹胀或腹泻等,部分患者起病隐匿,无任何症状。作为慢性胰腺炎的一种,AIP常见并发症有:糖尿病、门脉高压、胰腺结石、胰腺囊肿和胰腺萎缩。同时也常伴有IgG4-RD的胰腺外表现,如IgG4相关硬化性胆管炎、IgG4相关涎腺炎、IgG4相关肾病或IgG4相关肾病泪腺炎,此外也有血管炎、肝炎和血管炎等[3,7-8]。
2.IgG4相关硬化性胆管炎
IgG4相关硬化性胆管炎 (immunoglobulin G4 associated sclerosing cholangitis,IgG4-SC),又称为IgG4相关胆管炎(immunoglobulin G4 associated cholangitis,IAC),由 BjÖrnsson等[9]于2007年首次提出,是IgG4-RD在胆道系统的表现。美国2008提出了IgG4-SC的HISORt诊断标准[10],但我国常采用日本2012年推出的IgG4-SC诊断标准(表2)[11]。
与AIP类似,IgG4-SC患者多为老年男性,常见症状以梗阻性黄疸、腹部不适、脂肪泻和体重下降为主[12]。文献报道,大约20%~90%的IgG4-SC患者同时存在AIP[13-14],同时也可伴随其他IgG4-RD并表现出相应症状。
3.IgG4相关性肝病
IgG4相关性肝病 (immunoglobulin G4 associated hepatopathy)相对少见,以肝内炎性假瘤和肝功能异常为主要表现,同时可能伴有自身免疫性肝病 (autoimmune hepatitis,AIH)、原发性胆汁性胆管炎 (primary biliary cholangitis,PBC)、嗜酸粒细胞增多症、AIP或IgG4-SC等表现,目前仅以个案报道为主,天津医科大学总院于2017年进行了详细的综述[15]。

表1 美国2006年AIP诊断标准(基于HISORt标准)

表2 日本2012年IgG4-SC诊断标准
四、讨论
1.诊断与鉴别诊断
本例患者病例特点为老年男性,急性病程,以黄疸起病,伴纳差、体重下降,中间出现发热、腹痛等症状,影像学提示胰头占位、肝内外胆管扩张、肝内占位、多发淋巴结肿大、腹水及胸水,血清学提示肝功能异常、IgG4水平明显升高。A院已于疾病初发时诊断自身免疫性胰腺炎伴硬化性胆管炎,但未持续予激素治疗。B院因腹痛、发热及腹水考虑“消化道穿孔”行剖腹探查(具体不详,但我科胃镜检查未见消化道修补表现),随后肝占位表现被B院及我院误诊为“原发性肝癌”可能。我科入院后影像学初步结果仍提示“胆管细胞CA与肝脓肿鉴别”,但经肝占位活检及胆总管下段活检后证实IgG4-RD诊断。
血清IgG4水平升高可以作为IgG4-RD的线索,但超过7成的患者如原发性硬化性胆管炎 (primary sclerosing cholangitis,PSC)、胰腺炎、恶性肿瘤、肝硬化、AIH、炎症性肠病、系统性红斑狼疮及肝炎等也有IgG4水平升高,并且大约2成的确诊IgG4-RD患者IgG4水平正常[16]。因此,单一血清IgG4水平升高不能作为IgG4-RD的诊断依据,患者需要进一步检查。
影像学可以提供IgG4-RD诊断的更多线索,同时可以与相关疾病进行鉴别。AIP典型表现是胰腺弥漫性或局限性增大(腊肠样改变),较长的胰管狭窄但不伴远端扩张,但也只有3-5成患者具有典型表现,需要与胰腺癌、胆管癌及其他慢性胰腺炎相鉴别[3,17]。IgG4-SC典型表现是节段性胆管狭窄、胆管壁环腔均匀增厚,但确诊意义不大,需与胆管癌、PSC及其他非肿瘤疾病相鉴别[18-19]。 IgG4相关性肝病目前尚无典型表现总结,详细见上述及相关文献[15]。腹部超声及造影、CT、MRI、MRCP、IDUS、EUS 及 ERCP 等检查都可以帮助诊断。需要指出的是,影像学发现的多器官累及可以帮助诊断。
虽然组织中IgG4阳性浆细胞大量浸润是IgG4-RD的病理特征,然而确定诊断还应注意与其他肝胆胰疾病鉴别,如PSC、肝内炎性假瘤及滤泡性胆管炎等。同时,对IgG4免疫组化染色结果的解读要谨慎,因为恶性肿瘤及PSC中也会有IgG4阳性浆细胞浸润,现有的标准(>10个/HPF)敏感度较高,但特异度仍不足4成;组织IgG4阳性细胞 /IgG阳性比值>50%时敏感度和特异度较高,但是与血清学IgG4浓度变化无一定相关性[20]。
国际及国内共识均提供了诊断与鉴别诊断流程,可以参考进行诊断[6,11]。
2.诱导缓解和维持治疗
IgG4-RD的治疗一般参考AIP治疗建议,首选口服激素治疗,如激素疗效不佳,首先需要考虑诊断是否正确,然后可换用或联用免疫调节剂乃至利妥昔单抗。一般不建议手术治疗,当临床难以排除恶性肿瘤时可考虑手术。
IgG4相关疾病诊断及治疗国际专家共识推荐意见:激素多采取口服泼尼松 30-40mg/d(或 0.67 mg·kg-1·d-1),维持治疗2~4周后视临床效果逐渐减量,每2周减10~20 mg;20 mg/d维持2周后每2周减5 mg[21]。
需要指出的是,该治疗方案仍然存在争议。部分单位认为可以3~6月内停用激素,同时起始联合免疫抑制剂;但日本专家建议小剂量激素2.5~5 mg/d维持3年,且不需起始使用免疫制剂。我国国内也有不同意见[22]。
对于多器官受累、血清IgG4明显升高、累及近端胆管或既往曾有复发的患者,在诱导治疗达到临床缓解后,共识建议进行维持治疗[21]。
五、总结
通过该病例的诊治过程,我们的体会是在IgG4水平升高的线索上,完善影像学检查,充分鉴别诊断,重视活检病理及免疫组化的价值,积极早期地应用激素治疗并密切随访,可以明确诊断同时获得良好的治疗效果。
IgG4-RD作为新近发现的疾病,病因及机制尚未清楚,诊断标准和治疗方案尚在完善,仍需进一步的研究,特别是国内临床研究的基础上提出适合中国人群的诊断和治疗指南。
[1]Mahajan VS,Mattoo H,Deshpande V,et al.IgG4-related disease[J].Annual Review of Pathology,2014,9:315-347.
[2]张盼盼,赵继志,王木,等.IgG4相关性疾病346例临床特征分析[J].中华内科杂志,2017,56(9):644-649.
[3]Hart PA,Zen Y,Chari ST.Recent Advances in Autoimmune Pancreatitis[J].Gastroenterology,2015,149(1):39-51.
[4]Chari ST,Smyrk TC,Levy MJ,et al.Diagnosis of autoimmune pancreatitis:the Mayo Clinic experience[J].Clin Gastroenterol Hepatol,2006,4(8):1010-1016.
[5]Shimosegawa T,Chari ST,Frulloni L,et al.International consensus diagnostic criteria for autoimmune pancreatitis:guidelines of the International Association of Pancreatology[J].Pancreas,2011,40(3):352-358.
[6]《中华胰腺病杂志》编委会.我国自身免疫性胰腺炎共识意见(草案2012,上海)[J].中华胰腺病杂志,2012,12(6):410-418.
[7]赖雅敏,吴东,杨红,等.1型自身免疫性胰腺炎的流行病学及临床特点[J].基础医学与临床,2017,37(11):1607-1610.
[8]孙丽媛.自身免疫性胰腺炎诊断标准及诊疗手段研究进展[J].疑难病杂志,2017,16(9):955-958.
[9]BjÖrnsson E,Chari ST,Smyrk TC,et al.Immunoglobulin G4 associated cholangitis: description of an emerging clinical entity based on review of the literature[J].Hepatology,2007,45(6):1547-1554.
[10]Ghazale A,Chari ST,Zhang L,et al.Immunoglobulin G4-associated cholangitis:clinical profile and response to therapy[J].Gastroenterology,2008,134(3):706-715.
[11]Ohara H,Okazaki K,Tsubouchi H,et al.Clinical diagnostic criteria of IgG4-related sclerosing cholangitis 2012[J].J Hepatobiliary Pancreat Sci,2012,19(5):536-542.
[12]李雨涵,向晓星.IgG4相关性硬化性胆管炎的研究现状[J].临床肝胆病杂志,2017,33(11):2239-2242.
[13]Nakazawa T,Naitoh I,Hayashi K,et al.Diagnostic criteria for IgG4-related sclerosing cholangitis based on cholangiographic classification[J].J Gastroenterol,2012,47(1):79-87.
[14]Joshi D,Webster GJ.Biliary and hepatic involvement in IgG4-related disease[J].Aliment Pharmacol Ther,2014,40(11-12):1251-1261.
[15]张跃,周璐,王邦茂,等.免疫球蛋白G4相关性肝损伤的研究进展[J].中华消化杂志,2017,37(12):869-872.
[16]Culver EL,Sadler R,Simpson D,et al.Elevated Serum IgG4 Levels in Diagnosis,Treatment Response,Organ Involvement,and Relapse in a Prospective IgG4-Related Disease UK Cohort[J].Am J Gastroenterol,2016,111(5):733-743.
[17]Aravind S,Charisuresht,王槐志.自身免疫性胰腺炎[J].中华消化外科杂志,2011,10(5):325-328.
[18]杨素行,王屹.非肿瘤性疾病致梗阻性黄疸的影像学特征及鉴别诊断[J].中华消化外科杂志,2017,16(4):423-429.
[19]李燕妮,周璐,赵新,等.原发性硬化性胆管炎与IgG4相关硬化性胆管炎的异同[J].中华消化杂志,2017,37(3):213-216.
[20]袁农,毛羽丰.IgG4相关性肝胆胰疾病的病理学改变[J].中国医刊,2017,52(3):17-21.
[21]Khosroshahi A,Wallace ZS,Crowe JL,et al.International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease[J].Arthritis Rheumatol,2015,67(7):1688-1699.
[22]季兰岚,张卓莉.2015年IgG4相关疾病诊断及治疗国际专家共识推荐意见解读[J].中国实用内科杂志,2017,37(4):301-302.
猜你喜欢
中国临床医学影像杂志(2021年5期)2021-08-13 09:01:38
天津医科大学学报(2021年3期)2021-07-21 09:04:00
肝博士(2020年5期)2021-01-18 02:50:16
中国医学装备(2016年6期)2016-12-01 06:44:45
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:52
实用皮肤病学杂志(2015年4期)2015-12-22 11:21:40
肿瘤影像学(2015年3期)2015-12-09 02:38:50
医学研究杂志(2015年9期)2015-07-01 17:28:12
河南医学研究(2014年7期)2014-02-27 14:53:32
中国中西医结合外科杂志(2013年3期)2013-03-11 20:05:01
