
异喹啉酰肼构筑锌配合物的合成、晶体结构及其性质
2018-05-05 06:22吴文士黄妙龄石凤湘
无机化学学报 2018年5期
巴 聃 吴文士 黄妙龄 石凤湘
(华侨大学材料科学与工程学院,厦门 361021)
金属有机配合物的研究十分活跃,它们结构的多样性和性能的优越性,在多相催化、空腔隧道、磁性材料、非线性光学、抗癌抗菌药物和荧光等方面都有广泛的研究和应用[1-5]。异喹啉衍生物是一类典型的生物碱,作为一种重要的氮杂环化合物,广泛分布于现有的药物、功能材料以及其他各种生物体中[6]。 N,N′-乙酰异喹啉甲酰肼(Heiqnthz)和 N,N′-丙酰异喹啉甲酰肼(H2piqnthz)属于异喹啉衍生物,配体上的氮原子和氧原子是一种电子供体,可以与锌、镍等过渡金属离子形成螯合配合物。Znギ离子是d10型的副族元素,形成的配合物配位数可为4或6,几何构型可为四面体、三角双锥、四角锥、八面体等[7]。Znギ离子不仅拥有灵活多变的配位数和配位几何环境,而且与有机配体配位后,能展现出优异的荧光性能[8]。目前异喹啉衍生物与过渡金属形成的配合物大部分的应用研究集中在生物医药,尤其是抗癌与抗菌药物方面[9-10],例如锌与异喹啉衍生物的配合物在生物细胞毒性等方面的研究[11]。但是在荧光材料方面的研究未见报道。本文报道配合物[Zn(eiqnthz)2]·DMF(1)和配合物[Zn3(piqnthz)2Cl2]·2H2O(2)的晶体结构,并对其性质进行研究。
1 实验部分
1.1 材料和方法
原料药品主要购自北京百灵威公司,异喹啉-1-甲酸、水合肼(85%)、乙酸酐、丙酸酐、氟硼酸锌、氯化锌以及其他试剂均为分析纯,所有使用的试剂均未进行进一步提纯。用德国Elmentar Vario EL元素分析仪对碳、氢、氮元素进行元素分析。X射线粉末衍射数据在Smar/SmartLa粉末衍射仪上收集,辐射源为 Cu Kα(λ=0.154 056 nm),工作电压 40 kV,工作电流 30 mA,扫描角度(2θ)范围 5°~50°,扫描速度10°·min-1。在傅立叶变换红外光谱仪NICOLET iS50上进行红外测试,实验采用KBr压片,常温32次扫描测定红外光谱,波段范围为4 000~400 cm-1。用岛津DTG-60H热重分析仪,在氩气气氛条件下从26℃开始以10℃·min-1升温的速率,升温到1 000℃。用Lumiam分子光谱-赛默飞世尔Thermo荧光光度计进行荧光光谱分析。
1.2 有机配体Heiqnthz和[Zn(eiqnthz)2]·DMF(1)的合成
异喹啉甲酰肼的合成步骤完全参照文献[12]。在冰浴条件下,将制得的异喹啉-1-甲酰肼(0.10 mmol,21.71 mg)溶于二氯甲烷(20 mL)中与乙酸酐(0.10 mmol,1.1 mL)在磁力搅拌器下搅拌 30 min,然后室温条件下反应20 h。进行减压蒸馏和洗涤后得到白色固体粉末,用二氯甲烷和乙醇进行重结晶,产物Heiqnthz产率为82%。m.p.84℃,元素分析按C12H11N3O2计算值(%):C,62.87;H,4.84;N,18.33。实验值 (%):C,62.81;H,4.86;N,18.23。1H NMR(500 MHz,CDCl3):δ 10.47(s,1H),9.40(d,J=8.7 Hz,1H),9.08(s,1H),8.47(d,J=5.5 Hz,1H),7.84(dd,J=16.0,6.8 Hz,2H),7.70(dt,J=14.1,7.5 Hz,2H),2.18(s,3H)。 称 量Heiqnthz(0.01 mmol,2.31 mg) 和 氟 硼 酸 锌(0.01 mmol,1.27 mg)加到 DMF 和甲醇(1∶2.5,V/V)的混合溶液中,搅拌30 min,静置,自然挥发1周左右得浅黄色柱状晶体,合成产率为75%。元素分析按C27H27N7O5Zn 计算值(%):C,54.51;H,4.57;N,16.48。实验值(%):C,54.56;H,4.51;N,16.43。
1.3 有机配体H2piqnthz和{[Zn3(piqnthz)2Cl2](H2O)2}(2)的合成
在冰浴条件下,将制得的异喹啉-1-甲酰肼(0.10 mmol,21.71 mg)溶于二氯甲烷(20 mL)中与丙酸酐(0.10 mmol,1.5 mL)在磁力搅拌器下搅拌 30 min,室温条件下反应24 h。将所得的白色细针状晶体用三氯甲烷和甲醇的混合溶液进行重结晶,产物H2piqnthz产率为88%。m.p.142~143℃。元素分析按 C13H13N3O2计算值(%):C,64.19;H,5.39;N,17.27。实验值 (%):C,63.89;H,5.23;N,17.32%。1H NMR(500 MHz,CDCl3):δ 10.54(s,1H),9.39(d,J=8.7 Hz,1H),9.20(s,1H),8.45(d,J=5.5 Hz,1H),7.81(dd,J=20.9,6.8 Hz,2H),7.68(dt,J=15.5,7.0 Hz,2H),2.43(q,J=7.6 Hz,2H),1.24(t,J=7.6 Hz,3H)。 将 Heiqnthz(0.01 mmol,2.17 mg)和氯化锌(0.01 mmol,1.36 mmol)分别加到DMF和甲醇(1∶3,V/V)的混合溶液中进行搅拌 30 min,静置,自然挥发2周后得黄色柱状晶体,合成产率为80%。元素分析按C26H31Cl2N6O9Zn3计算值(%):C,37.24;H,3.73;N,10.02;实验值(%):C,37.29;H,3.70;N,10.11。
1.4 单晶X射线衍射实验
选择尺寸为0.09 mm×0.09 mm×0.08 mm的配合物单晶1和0.06 mm×0.05 mm×0.04 mm的配合物单晶2。用Bruker Smart Apex CCD X射线单晶衍射仪,经石墨单色器单色化的λ=0.071 073 nm的Mo Kα为辐射光源。在293(2)K条件下以φ-ω方式收集衍射数据,数据通过经验吸收校正。选取I>2σ(I)的可观察点用于结构解析,结构解析和精修采用SHELXL-97[13]。配合物1和2的晶体数据和结构的精修数据列于表1,而部分键长键角列于表2。
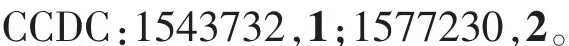

表1 配合物1和2的晶体数据及其精修参数Table 1 Crystal data and refinement parameters of complexes 1 and 2
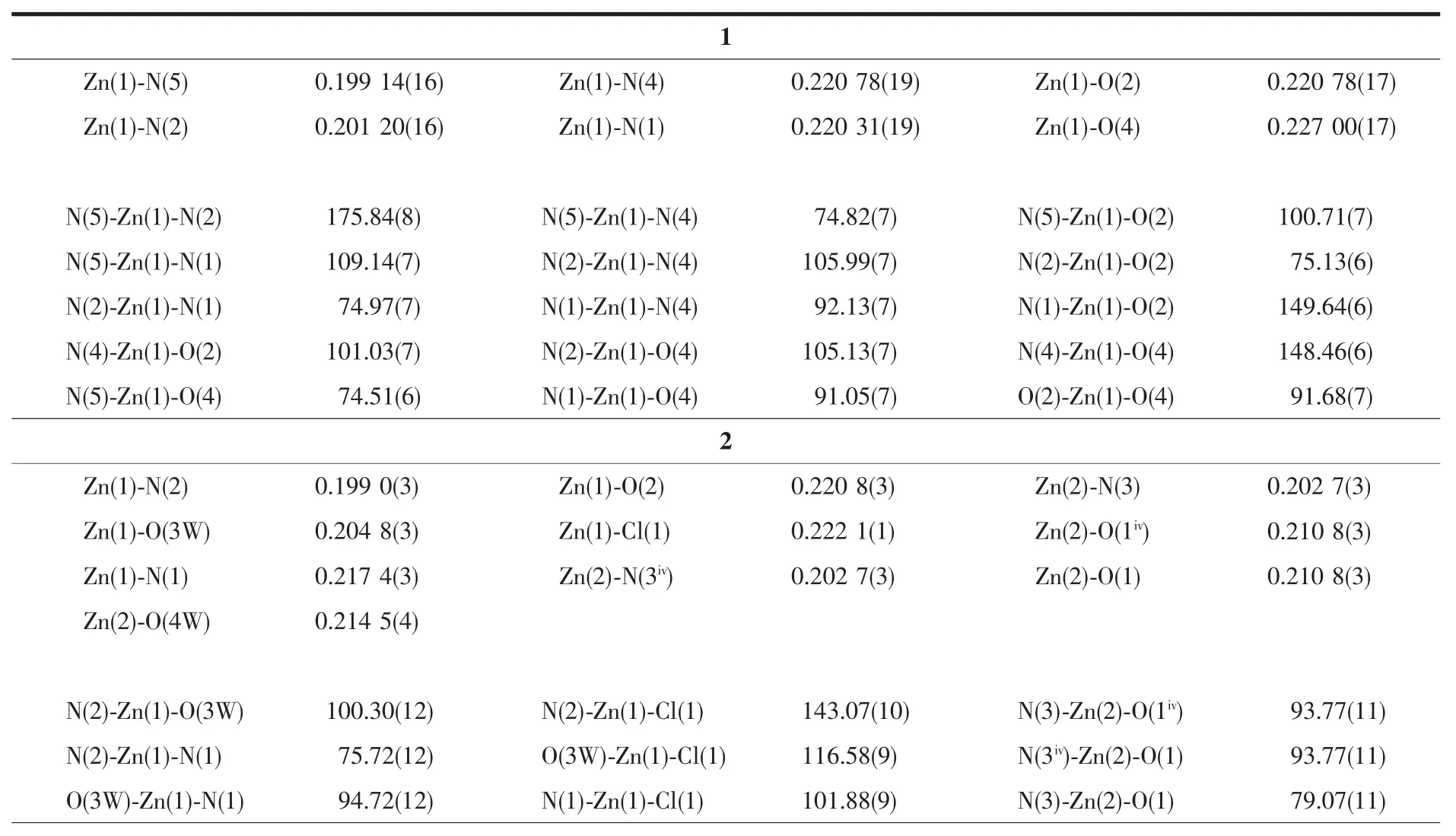
表2 配合物1和2的部分键长(nm)和键角(°)Table 2 Selected bond lengths(nm)and bond angles(°)for the complexes 1 and 2

续表1
2 结果与讨论
2.1 配合物1的晶体结构

图1 配合物1的结构图:(a)分子结构图;(b)一维链氢键结构图和多面体图;(c)π-π堆积图;(d)三维分子结构图Fig.1 Structure of complex 1:(a)Molecular structure;(b)Structure of hydrogen bonds and polyhedral graphs;(c)π-π stacking;(d)3D structure
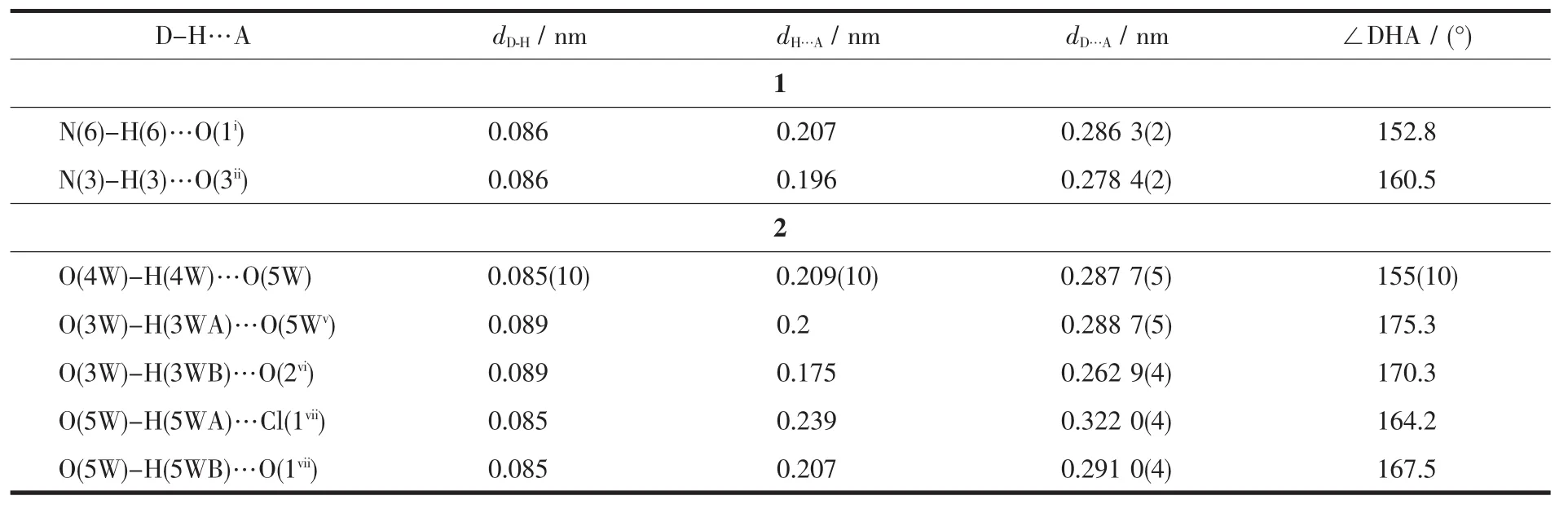
表3 配合物1和2的氢键参数Table 3 Hydrogen bond parameters for complexes 1 and 2
单核金属配合物1属于单斜晶系,C2/c空间群。如图1a所示,锌ギ离子与周围原子配位呈扭曲的 N4O2八面体构型,4 个配位原子 N(1)、N(4)、O(2)和O(4)与Zn(1)配位形成赤道平面,键长分别为0.220、0.201、0.221、0.227 nm,N(2)、N(5)分别处于 2个轴向位置,键长分别为0.201、0.199 nm,均属于正常的 Zn-N 配位键长范围。 N(1)、N(4)、O(2)和 O(4)原子与赤道最小二乘平面间的距离偏差为0.058 nm,共面性好,锌与赤道平面共面。N(4)在赤道平面上方,O(4)在赤道平面下方,N(2)-Zn(1)-N(5)的键角为175.8°,与直线偏离仅4.2°,两极几乎垂直于赤道平面。在分子中2个配体L1、L2与Zn(1)配合物形成4个五元螯合环 M1(N(1)、C(1)、C(10)、N(2)、Zn(1),各原子与其最小二乘平面的距离偏差值Rms=0.008 nm);M2(N(2)、N(3)、C(12)、O(2)、Zn(1),Rms=0.039 nm);M3(N(5)、N(6)、C(23),O(4),Zn(1),Rms=0.009 nm);M4(N(4)、C(13)、C(22)、N(5)、Zn(1),Rms=0.031 nm)。 配体 L1与Zn(1)形成的螯合环M1、M2的二面角为6.53°,配体L2与Zn(1)形成的螯合环M3、M4的二面角为5.50°,各螯合环共面性均很好。配合物分子之间通过分子间氢键(N(6)-H(6)…O(1i)和N(3)-H(3)…O(3ii)的相互作用形成一维无限链状结构(图1b、表3),氢键键长(dD…A)分别为0.286和0.278 nm,属于典型的氢键。相邻链间,异喹啉环与异喹啉环部分重叠,存在 π-π 堆积(图 1c):π 平面(C(13),N(4),C(14),C(15),C(16),C(17),C(18),C(19),C(20),C(21), 质 心 Cg(1)(0.71257,0.85244,0.42036))与相邻 π 平面(C(13iii),N(4iii),C(14iii)C(15iii),C(16iii),C(17iii),C(18iii),C(19iii),C(20iii),C(21iii),质心 Cg(2iii)(0.787 43,0.647 56,0.579 64))的二面角为0.09°,2个π平面近似平行,质心间距离为0.413 18 nm,平面间垂直距离为0.338 62 nm,平面相对滑移0.245 79 nm。此π-π堆积作用使相邻两条链结合更牢固。π-π堆积和氢键对配合物3D空间结构的稳定性起到关键作用(图1d)。
2.2 配合物2的晶体结构
配合物2属于单斜晶系,C2/c空间群,三核结构。如图2a所示,三核金属配合物整体呈现V字型,Zn(1),Zn(3)位于分子两端并且是变形四角锥配位构型,Zn(2)位于中间形成三角双锥配位构型(图2b)。反对称结构单元由3个Znギ离子,2个Cl-离子,2个阴离子配体piqnthz2-(简写为L3,L4)以及3个水分子构成。Zn(1)离子周围有2个N原子,2个O 原子以及 1个 Cl-与之配位,其中 N(1)、N(2)、O(2)来自配体L3,O(3W)由配位水分子提供。这5个配位原子在Zn(1)离子周围形成变形四角锥形(C4v)。Cl(1),O(2),N(2),N(1)原子在四角锥的底面 4 个顶点处,最小二乘平面间的距离偏差为0.020 nm。配位水中的氧原子(O(3W))处于四角方锥的顶点。Zn(2)离子分别与来自L3中的氮原子N(3)和氧原子O(1)以及L4中的氮原子N(3iv)和氧原子O(1iv)配位,另外还有一个氧O(4W)原子是由配位水分子提供,这5个原子形成三角双锥(D3h)几何构型。3个氧原子O(1),O(4W),O(1iv)共平面,形成三角双锥的赤道平面。N(3)和N(3iv)分别处于三角双锥的顶点位置,形成赤道平面的两极,N(3)和N(3iv)到平面垂直的距离都为0.199 nm,N(3)-Zn(2)的键长为 0.203 nm,N(3)-Zn(2)-N(3iv)为 166.49(18)°,偏离直线达 13.51°,轴与赤道平面接近垂直。Zn(3)与来自配体L4中的N(1iv),N(2iv),O(2iv),以及配位水分子中的 O(3Wiv)和 Cl-进行配 位 形 成 变 形 四 角 锥 形(C4v)。 Cl(1iv),O(2iv),N(2iv),N(1iv)这4个原子在四角锥底面的4个顶点处,配位水中的氧原子O(3Wiv)处于四角方锥的顶点处。
该配合物中存在丰富的氢键,相邻配合物之间通过氢键(O(4W)-H(4W)…O(5W);O(3W)-H(3HA)…O(5Wv);O(5W)-H(5WB)…O(1viii))相互作用形成鱼鳞形的一维链(图2c),再通过O(5W)-H(5WA)…Cl(1vii),O(3W)-H(3HB)…O(2vi)拓展形成二维结构(图 2d,表3)。π 平面(C(1),N(1),C(2),C(3),C(4),C(5),C(6),C(7),C(8),C(9),质心 Cg(1)(0.749 97,0.506 93,0.495 50))与相邻 π 平面(C(1ix),N(1ix),C(2ix),C(3ix),C(4ix),C(5ix),C(6ix),C(7ix),C(8ix),C(9ix)), 质 心 Cg(2ix)(0.749 97,-0.006 93,0.504 50))的二面角为 0.13°,质心间距离为0.390 51 nm,2个π平面间垂直距离为0.343 69 nm,平面相对滑移为0.185 41 nm。说明分子间产生错位π-π堆积效应(图2e)。这些π-π堆积将相邻的分子链相连接,把配合物拓展成为3D网络分子结构(图 2f)。
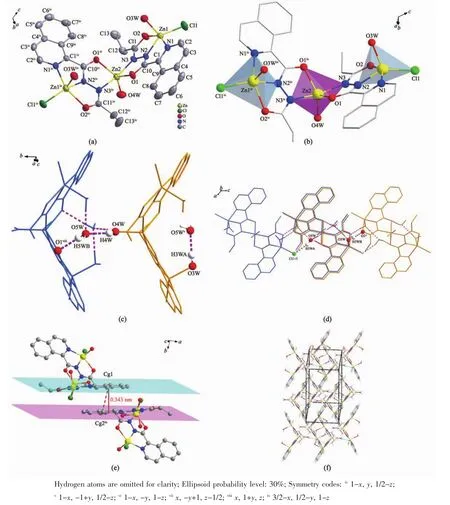
图2 配合物2的结构图:(a)分子结构图;(b)多面体图;(c)氢键连接形成的一维链结构图;(d)氢键连接形成的二维结构图;(e)π-π堆积图;(f)三维结构图Fig.2 Structure of complex 2:(a)Molecular structure;(b)Polyhedral graphs;(c)1D structure connected by hydrogen bonds;(d)2D structure connected by hydrogen bonds;(e)π-π stacking;(f)3D structure
2.3 红外光谱分析
红外图谱分析表明,Heiqnthz的特征吸收峰主要在3 355,3 039,2 926,1 693,1 659,1 583,1 504,1 423,1 452,1 377 cm-1。形成配合物1后,在1 704 cm-1出现肽胺羰基特征吸收峰,而1 622,1 568,1 546,1 427 cm-1处的较强特征吸收峰主要是喹啉环的骨架振动,2 907,1 423,1 377 cm-1的 3 个峰分别为-CH3的C-H伸缩振动,-CH3对称伸缩振动以及反对称伸缩振动。与Heiqnthz相比配合物1出现1 672 cm-1的红移峰,在3 160 cm-1处出现酰胺的N-H伸缩振动峰,峰位发生蓝移,这可能与烷酰胺与金属发生配位有关。Heiqnthz与H2piqnthz结构相似,峰位置基本相似,H2piqnthz与金属形成配合物2时配位点与配合物1配位点相同,因此峰位置也相似。但配合物2在3 445 cm-1处有强的吸收峰,表明配合物2中存在结晶水。
2.4 配合物热稳定性分析及其固体粉末衍射分析
从TGA曲线可以看出(图3a),配合物1在40℃左右开始出现了DTA热吸收峰,这意味着配合物可能存在游离水,从323℃开始失重到890℃左右曲线趋近于平坦,残重12.5%(理论值13.7%)。配合物2的TG曲线(图3b)主要分为2段,第一段从159℃左右开始分解到189℃左右,主要是失去2个结晶水,大约失重4.96%,理论值为4.29%。下一阶段从223℃开始主要出现结构的坍塌和分解,到578℃曲线开始趋于平坦,表明样品分解基本完全,残物为ZnO(实验值26.53%,理论值28.59%)。由此分析配合物1和2结构的热稳定性比较好。
在室温条件下,对配合物1和2进行了X射线粉末衍射表征。如图4所示,配合物1的模拟峰与实验测定的峰值几乎一致(图4a),表明该配合物纯度比较高。配合物2的模拟峰和实验测定的峰基本吻合,但是峰强度存在一定的差异(图4b)。

图3 配合物1(a)和2(b)的TG-DTA曲线Fig.3 TG-DTA curves of complexes 1(a)and 2(b)

图 4 配合物 1(a)和 2(b)的 PXRD 图Fig.4 PXRD patterns of complexes 1(a)and 2(b)
2.5 荧光光谱分析
在室温下,对 Heiqnthz、配合物 1、H2piqnthz 和配合物2这4种固体进行荧光光谱分析。从图5a、b分析可知,Heiqnthz在波长 λem=440.6 nm(λex=372.2 nm)(蓝色光)和配合物1在波长λem=494.3 nm(λex=360.3 nm)处有较大的发射峰,配合物1相对于Heiqnthz荧光发射波长红移了54.3 nm,从蓝光波段红移到了青光波段。在图5c、d中H2piqnthz在波长 λem=478.6 nm(λex=372.2 nm)和配合物 2 在波长λem=560.5 nm(λex=494.3 nm)处发现有较大的发射峰。与H2piqnthz相比配合物2发生了较大的红移(81.9 nm)。在相同的激发波长下,配体H2piqnthz和Heiqnthz相比发射波长也发生红移(38 nm)。可能是H2piqnthz增加1个-CH3基团对结构的吸供电子共轭体系产生一定的影响。配合物2和配合物1相比红移更大,是因为配合物2两个配体之间的二面角为117.75°,而配合物1的二面角为94.51°。配合物2的刚性增强,导致红移更大。荧光的产生主要有3种途径:金属离子微扰的配体的荧光,配体微扰的金属离子荧光,以及金属离子到配体电荷转移跃迁(MLCT)的发光。锌离子是一种典型的d10金属价电子结构,与有机配体发生配位展现出荧光性能。配合物1和2的荧光都是金属到配体电荷迁移(MLCT)导致的。

图5 配体与配合物的荧光发射光谱Fig.5 Fluorescence emission spectra of the ligands and complexes
利用Guassian03程序,以配合物以及配体的晶体数据作为分子模型,以3-21G为标准基组,运用HF(Hartree-Fork)从头算法对配合物进行理论计算。配体Heiqnthz和配合物1,配体H2piqnthz及其配合物2的电荷及其自旋多重度都是取(0,1)。配体1与Zn形成配合物后极性从2.150 7变为2.178 7,主要键的Mulliken重叠布居数均在0.07以上,配位后配体2总偶极矩从2.108 2变为5.248 4。配体形成配合物后的对称性大大降低了。计算得到前线轨道:配合物 1:EHOMO=0.269 32 a.u.,ELUMO=0.010 62 a.u.,ΔE=0.279 94 a.u.(图 6a);配合物 2:EHOMO=0.260 92 a.u.,ELUMO=-0.022 18 a.u.,ΔE=0.283 1 a.u.(图 6b)。 配合物1与Heiqnthz相比,轨道能隙ΔE是降低的,配合物2由LUMO轨道跃迁到HOMO轨道所放出的能量值也较H2piqnthz的值低,可见配合物较之配体荧光发生红移(表4),这与荧光谱图的结果基本吻合。

图6 配合物1(a)和2(b)的分子前线轨道图Fig.6 Frontier molecular orbitals of complexes 1(a)and 2(b)

表4 配体与配合物的轨道能Table 4 Orbital energy of the ligands and complexes
3 结 论
采用普通溶液法合成了配合物 [Zn(eiqnthz)2]·DMF(1)和[Zn3(piqnthz)2Cl2]·2H2O(2),通过 X 射线单晶衍射得到这2种配合物的晶体结构。配合物1和2的配体相似,拥有相同的空间群,但配位方式存在很大的差异。配合物1属于单核结构,配合物2属于三核结构,并且配合物2中存在四方锥和三角双锥2种几何构型。配合物1和2中丰富的氢键和ππ堆积作用,对配合物的三维结构形成和稳定有很大的作用。荧光分析发现配体在与锌形成配合物后发生荧光红移现象,表现出较好的荧光性能,是潜在的荧光材料。
参考文献:
[1]GUO Jian-Qiang(郭建强),SUN Yin-Xia(孙银霞),YU Bin(俞彬),et al.Chinese J.Inorg.Chem.(无机化学学报),2017,33(8):1481-1488
[2]Yoon M,Srirambalaji R,Kim K.Chem.Rev.,2012,112(2):1196-1231
[3]ZHANG Jie(张杰),TENG Ming-Yu(滕明瑜),DAI Guo-Liang(戴国梁),et al.Chinese J.Inorg.Chem.(无机化学学报),2014,30(3):499-505
[4]Ferrari M B,Bisceglie F,Pelosi G,et al.J.Inorg.Biochem.,2002,90(3/4):113-126
[5]Allendorf M D,Bauer C A,Bhakta R K,et al.Chem.Soc.Rev.,2009,38(5):1330-1352
[6]Bringmann G,Zhang G,Büttner T,et al.Chem.Eur.J.,2013,19(3):916-923
[7]Yan L,Li C B,Cui D,et al.Chin.J.Struct.Chem.,2015,34(2):221-226
[8]CHANG Hui-Qin(常慧琴),YUAN Zhi-Ze(原知则),LAI Xiao-Qing(赖晓晴),et al.Chinese J.Inorg.Chem.(无机化学学报),2016,32(11):2058-2062
[9]Bringmann G,Zhang G,Büttner T,et al.Chem.Eur.J.,2013,19(3):916-923
[10]Bermejo A,Andreu I,Suvire F,et al.J.Med.Chem.,2002,45(23):5058-5068
[11]Huang K B,Mo H Y,Chen Z F,et al.Eur.J.Med.Chem.,2015,100:68-76
[12]Yeoh K K,Chan M C,Thalhammer A,et al.Org.Biomol.Chem.,2013,11(5):732-745
[13]Sheldrick G M.Acta Crystallogr.Sect.A:Found.Crystallogr.,2008,A64:112-122
猜你喜欢
高中数理化(2022年14期)2022-08-15
波谱学杂志(2021年3期)2021-09-07
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
应用化工(2020年6期)2020-07-30
理化检验-化学分册(2020年4期)2020-06-03
原子与分子物理学报(2019年5期)2019-04-28
当代陕西(2019年6期)2019-04-17
中成药(2017年7期)2017-11-22
中学化学(2015年12期)2016-01-19
外语学刊(2014年3期)2014-12-03
