
溶剂热法合成Fe-CeO2与N-Fe-CeO2纳米粉体及其光催化性能
2018-05-05 06:22黄建平折小梅石惠民
无机化学学报 2018年5期
黄建平 陈 芳 折小梅 王 赫 石惠民
(1湖南大学,材料科学与工程学院,长沙 410082)
(2湖南大学,物理与电子科学学院,长沙 410082)
0 引 言
近年来,随着工业技术的发展,人民生活水平逐步提高,但环境污染问题也日益严峻。光催化技术是利用光能治理有机污染物、无机污染物和制取氢等的一门新兴技术,引起了学者的广泛关注。目前,除了技术相对成熟并且已获广泛应用的TiO2外,新型光催化剂也不断被开发出来。其中,CeO2由于具有良好的催化活性和稳定性,较高的氧化还原性能(Ce3+/Ce4+),且成本低、无毒等特点,在光催化领域的应用前景已倍受关注[1-3]。与TiO2的宽禁带,太阳能利用率低,量子效率低等缺点类似[4],CeO2的禁带宽度约3.2 eV[5],通常只吸收400 nm以下的紫外光,而不能充分利用太阳光中能量集中的可见光。因此唯有降低CeO2的带隙宽度,使其光响应范围延伸至可见光区才能从根本上提高CeO2光催化活性。掺杂是提高CeO2光催化活性的有效途径之一,通过适当的掺杂,在CeO2带隙中引入杂质能级以减小其带隙宽度。目前通过掺杂改善CeO2光催化性能的报道可分为2种:一种是掺杂过渡金属和稀土元素,如 Fe、Co、Y、La 等[5-12]。Wang 等[7]采用水热法、共沉淀法和溶剂热法3种方法合成了Fe掺杂CeO2纳米片,发现在100~175℃的水热合成的Fe掺杂CeO2对二氯乙烷的光催化性能最好,而175~350℃高温合成条件下,溶剂热法制备Fe掺杂CeO2催化性能更佳。袁强等[8]采用溶胶-凝胶法和浸渍法制备了Fe/CeO2固体催化剂,用来催化刚果红,其最高催化效率达96%。Arul等[9]采用溶剂热法合成了Fe掺杂CeO2,证实了其对亚甲基蓝的降解率得到提高,但并未指出Fe的最佳掺杂量。他们还合成了Co掺杂CeO2,结果证明Co-CeO2对偶氮染料的光催化性能良好[10]。Liyanage等[5]在CeO2中掺杂不同含量的Y,发现随着Y含量的增加,对靛蓝胭脂红、罗丹明B的光催化降解性能都逐步提高。另一种是掺杂非金属元素,如N、C等[13-19]。顾明杰等[14]以三乙醇胺为氮源,通过溶剂热法成功制备氮掺杂纳米CeO2,并且发现氮掺杂CeO2对酸性橙的降解率高达93.9%。Mao等[15]以乙二胺为氮源,通过蒸馏回流法成功将氮掺入CeO2,通过可见光催化分解亚甲基蓝溶液,发现CeO2掺杂4%氮时光催化活性最好。Wu等[16]以浓硝酸为氮源,采用溶剂热法将氮掺入CeO2,结果显示掺氮CeO2对罗丹明B的降解率是纯CeO2的12.6倍。翦立新等[19]利用微波等离子技术结合溶胶凝胶法制备了Fe3+-N-CeO2,发现当掺Fe量为0.1%时,Fe3+-N-TiO2具有最好的催化活性,而N掺杂进一步增强了对可见光的利用率。因此,通过适当的方式,掺杂适量的过渡金属、稀土金属或非金属元素,在一定程度上可提高CeO2的光催化性能。但由于金属元素与非金属元素在CeO2晶格中掺杂位置不相同,对光催化性能的提高机制也不相同。
因此,为了考察金属与非金属元素共同掺杂对CeO2光催化性能的协同效果,本文以Ce(NO3)3·6H2O为铈源,Fe(NO3)3·9H2O为铁源,制备了不同含量Fe掺杂CeO2(Fe-CeO2),并用不同含氮溶剂为氮源,制备了不同氮源的N-10%Fe-CeO2纳米粉体,并考察了其在模拟太阳光照条件下对有机污染物亚甲基蓝(MB)的光催化降解性能。
1 实验部分
1.1 主要试剂
所用的化学试剂主要有六水合硝酸铈 (威海佰德信新材料有限公司,分析纯,>98%),九水合硝酸铁(上海阿拉丁生化科技股份有限公司,分析纯,>98%),乙二醇(国药集团化学有限公司,分析纯,>99%),浓氨水(成都市科龙化工试剂厂,分析纯,24%~26%),尿素(天津市恒兴化学试剂制造有限公司,分析纯,>99%),三乙醇胺(上海阿拉丁生化科技股份有限公司,分析纯,>98%),二乙醇胺(上海阿拉丁生化科技股份有限公司,分析纯,>99%),亚甲基蓝(天津市恒兴化学试剂制造有限公司,指示剂,>98%)。氢氧化钠(天津市进丰化工有限公司,分析纯,>98%)。
1.2 样品的制备
1.2.1 Fe-CeO2催化剂的制备
以Ce(NO3)3·6H2O为铈源,乙二醇为溶剂,采用溶剂热法制备了不同掺杂含量的Fe-CeO2纳米粉体。首先按照总物质的量为0.01 mol,且nFe/(nFe+nCe)=0%,5%,10%,15%分别计算出 Ce(NO3)3·6H2O 和Fe(NO3)3·9H2O 的质量。 先将 Ce(NO3)3·6H2O 溶解于35 mL乙二醇搅拌至溶解后,再加入对应的Fe(NO3)3·9H2O,再搅拌30 min至充分溶解,再加入5%NaOH溶液,调节其pH值为9~10,继续搅拌30 min。将溶液转移至50 mL聚四氟乙烯内衬反应釜中,在180℃的马弗炉中反应24 h。然后随炉冷却至室温,取出生成的沉淀,用去离子水和乙醇溶液反复洗涤数遍,离心分离后放入70℃干燥箱中干燥。最后将干燥好的粉体500℃煅烧2 h,即得到不同物质的量比的Fe-CeO2纳米粉体。
1.2.2 N-Fe-CeO2催化剂的制备
以 Ce(NO3)3·6H2O 为铈源,Fe(NO3)3·9H2O 为铁源,三乙醇胺(TEA),氨水,尿素(urea),二乙醇胺(DEA)为氮源,按上述nFe/(nFe+nCe)=10%制备出N-10%Fe-CeO2粉体。首先称取4份3.9 g的Ce(NO3)3·6H2O溶于30 mL乙二醇中,再分别加入0.404 g的Fe(NO3)3·9H2O,搅拌30 min至完全溶解混合,分别加入10.7 mL的三乙醇胺,12 mL浓氨水,4.8 g尿素,7.7 mL二乙醇胺,继续搅拌30 min使其充分混合,将溶液转移至50 mL聚四氟乙烯内衬反应釜中,在180℃的马弗炉中反应24 h。样品的洗涤、干燥与煅烧过程与Fe-CeO2粉体一致。样品分别标记为 TEA-N-10%Fe-CeO2,NH3·H2O-N-10%Fe-CeO2,Urea-N-10%Fe-CeO2,DEA-N-10%Fe-CeO2。
1.3 样品的表征
采用D/Max2500型X射线衍射仪(XRD)分析了样品的物相成分,管电压为40 kV,电流为10~450 mA, 靶材为铜靶(λ=0.154 nm), 扫描范围为20°~80°;采用JEM-2100型透射电子显微镜(TEM)观察了样品的微观形貌,加速电压200 kV;采用ORIUS SC1000能量散射X射线光谱分析仪(EDS)分析了样品的元素分布状态。采用ESCALAB 250Xi型X射线光电子能谱(XPS)对样品的表面化学状态进行了分析,X射线光源为200 W单色Al Kα射线;采用LabRAM-010激光拉曼光谱仪对样品进行拉曼分析,激发波长为532 nm;采用Agilent Cary 300紫外-可见分光光度计(UV-Vis)测试固体样品的吸光度。
1.4 粉体的光催化性能及循环使用性能测试
取50 mg光催化剂分散于50 mL浓度为10 mg·L-1的亚甲基蓝(MB)溶液,先在搅拌作用下暗反应30 min,以达到吸附脱附平衡。然后将溶液置于氙灯(PLS-Microsolar 300 W)下照射,持续不断电磁搅拌,光源距溶液表面10 cm,光功率密度约为2 000 mW·cm-2。每隔30 min取样,离心分离得上清液,用TU-1901紫外可见分光仪测试溶液在最大吸收波长664 nm的吸光度。由于吸光度与溶液浓度成正比,遵循朗伯-比尔定律,因此可用吸光度A/A0来表示溶液的浓度变化C/C0,从而评价光催化剂的催化效应。A,C分别为反应时间为t时的吸光度值和溶液浓度,A0,C0分别为暗反应前的吸光度值和溶液浓度。
将每次光催化试验后的催化剂,离心分离进行回收,使用去离子水反复洗涤多次,烘干后再次测试其光催化性能。如此多次循环测试,以验证光催化剂在循环使用时的催化性能的稳定性。
2 结果与讨论
2.1 TEM分析
图 1(a~d) 分别为纯 CeO2、10%Fe-CeO2、NH3·H2O-N-10%Fe-CeO2和Urea-N-10%Fe-CeO2的TEM图片。从图中可以看出当添加NaOH为沉淀剂,无氮源时,所得纳米颗粒具有较强的团聚性。而浓氨水为氮源时,所获粉体分散较好,许多粉体堆积形成中空薄壁泡沫状,而以尿素为氮源时,所获粉体表现为无序的草叶状。CeO2基材料的形貌对生长环境非常敏感,目前已合成了大量形貌各异的CeO2粉体[20]。本实验中掺杂CeO2形貌主要受pH值变化,氮源的特性及与溶剂的共同作用。根据其高分辨插图,测量了其晶面间距为0.306~0.315 nm之间,对应于萤石结构CeO2的(111)面。说明纳米颗粒的表面主要是以密排面(111)面为主。对NH3·H2ON-10%Fe-CeO2粉体进行了EDS能谱分析,发现N、Fe在粉体中的分布比较均匀,也证明了N、Fe在粉体中均匀掺杂。图1(e~h)分别为图1(c)中框内区域的Ce、Fe、N和O等元素的分布图。可以看出这些元素在粉体中均已存在且分布比较均匀。说明Fe、N等元素已均匀掺杂进入了CeO2晶格中。
2.2 XRD分析
利用X射线衍射仪对掺杂Fe、N-Fe的样品的晶体结构进行了分析,如图2所示。图2(a)为不同含量Fe掺杂CeO2的XRD图。图中的衍射峰均表现为CeO2的立方萤石结构的特征峰 (PDF No.34-0394)。图中没有其它特征峰,表明掺杂的Fe已全部进入CeO2晶格。随着掺杂Fe含量的增加,衍射峰的位置向高衍射角方向逐渐移动,说明掺杂使得其晶格常数减小。图2(b)为10%Fe-CeO2中加入了不同氮源合成的样品的XRD图,其衍射峰同样均表现为CeO2萤石结构的特征峰。衍射峰的位置,半高宽等外形特征基本相似。利用Scherrer公式和(111)、(200)、(220)、(311)的衍射峰的参数计算了晶粒尺寸,即:


图 1 (a)纯 CeO2、(b)10%Fe-CeO2、(c)NH3·H2O-N-10%Fe-CeO2 和(d)Urea-N-10%Fe-CeO2 的 TEM 图;(e~h)分别为图 1(c)中红框内的 Ce、Fe、N、O 的元素分布图Fig.1 TEM images of(a)pure CeO2,(b)10%Fe-CeO2,(c)NH3·H2O-N-10%Fe-CeO2and(d)Urea-N-10%Fe-CeO2;(e~h)Elements distribution of Ce,Fe,N and O for the area in frame in Fig.1(c)
式中,k为谢乐常数,取值0.89;λ为入射X射线波长,取值为0.154 nm;θ为衍射角;B为实测样品衍射峰半高宽度。利用布拉格衍射定律和面心立方结构晶面间距的计算公式计算了晶体的晶格常数,如表1所示。晶格常数随着掺杂Fe含量的增加不断减小,主要是由于半径较小的Fe3+(65 pm)取代了半径较大的Ce4+(97 pm)的位置。而不同的氮源对其氧化铈的晶格常数影响较小,主要是由于N3-(146 pm)离子半径与O2-(140 pm)半径接近。晶粒尺寸的变化不大,为10~12 nm之间。说明这种合成方法对晶粒尺寸分布的可控性强。
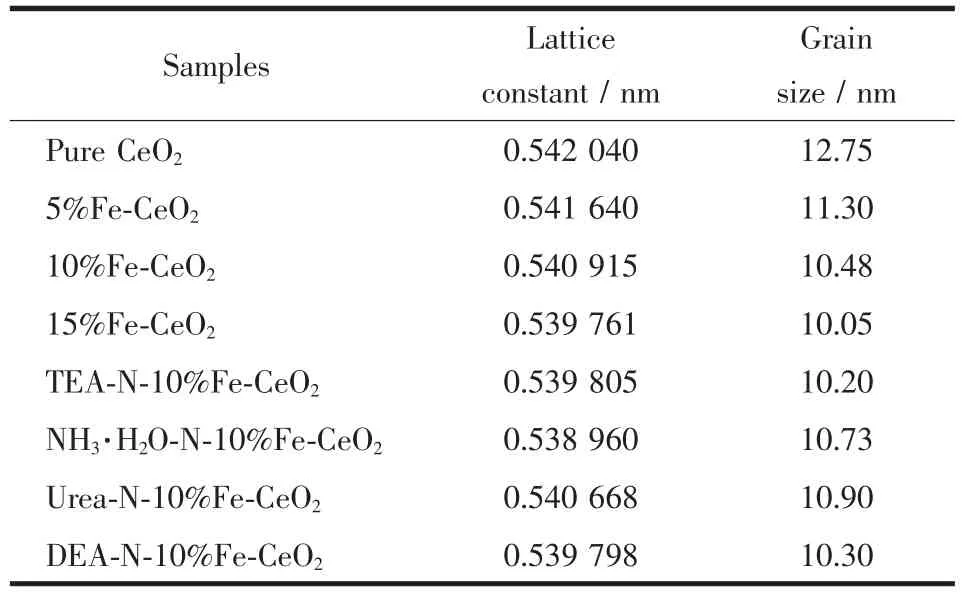
表1 样品的晶格常数与晶粒尺寸Table 1 Lattice constants and grain sizes of the samples

图 2 Fe-CeO2(a)与 N-10%Fe-CeO2(b)的 XRD 图Fig.2 XRD patterns of Fe-CeO2(a)and N-10%Fe-CeO2(b)
2.3 XPS分析
图 3 为 NH3·H2O-N-10%Fe-CeO2与 10%Fe-CeO2中Fe2p、Fe3p和N1s的XPS高分辨图谱。采用软件XPSpeak4.1对Fe2p XPS图谱进行分峰处理,如图3(a)所示。711.4和717.4 eV分别为Fe2p3/2的主峰及卫星峰;724.4和733.2 eV分别为和Fe2p1/2是结合能主峰及卫星峰。卫星峰的强度比主峰强度高说明Fe进入CeO2晶格后,Fe与Ce之间存在着很强的相互作用。图3(b)显示了Fe3p的位置约为 55.6 eV,主要对应为 Fe3+[21]。 图 3(c)中 399.2和402.9 eV分别代表N在CeO2晶格中和吸附的N2的结合能峰[18-19]。通过对比,容易看出,浓氨水掺杂后,CeO2晶格中成功掺杂了一定量的N。根据各元素结合能峰可估算出10%Fe-CeO2中nFe/(nFe+nCe)=9.2%,NH3·H2O-N-10%Fe-CeO2中 nFe/(nFe+nCe)=5.9%,nN/(nO+nN)=0.9%。
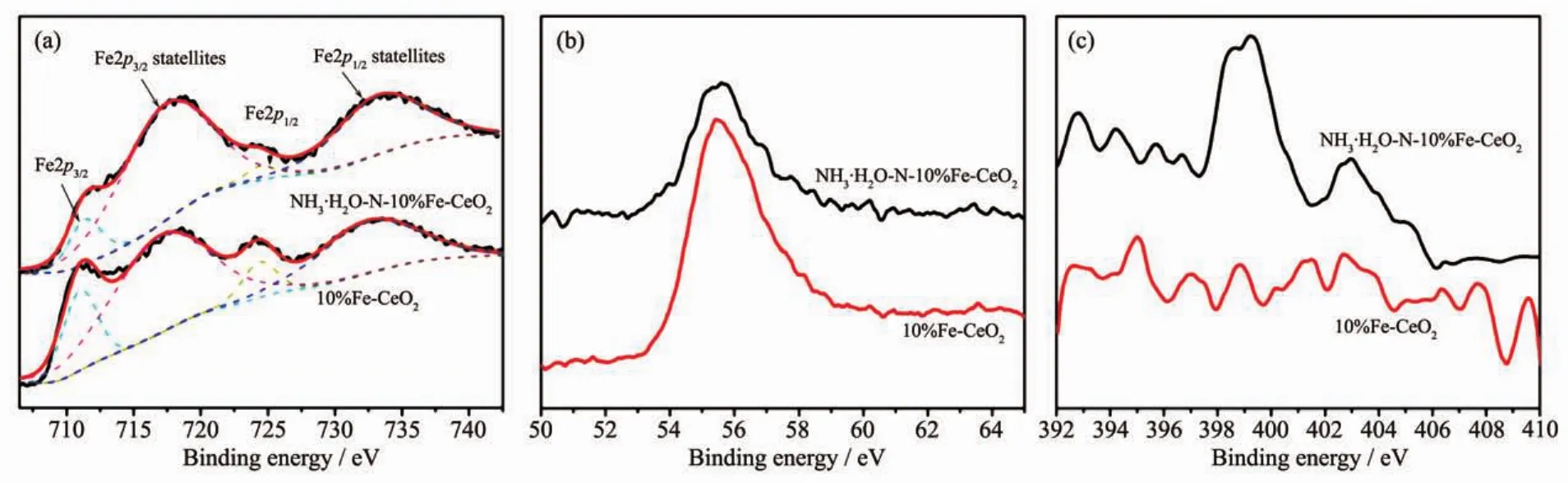
图 3 NH3·H2O-N-10%Fe-CeO2及 10%Fe-CeO2的 Fe2p(a)、Fe3p(b)和 N1s(c)的 XPS 图谱Fig.3 Fe2p(a),Fe3p(b)and N1s(c)XPS spectra of NH3·H2O-N-10%Fe-CeO2and 10%Fe-CeO2
2.4 Raman分析
图4 显示了不同含量Fe掺杂CeO2与不同氮源的N-10%Fe-CeO2的Raman图谱。纯CeO2的Raman图谱在462.4 cm-1处存在一个F2g模式振动峰。随着掺杂Fe含量的增加,F2g峰的位置稍有红移,强度也逐渐降低,随着掺杂Fe含量的增加,极化率逐渐降低,以致当Fe掺杂含量达到15%时,其F2g近乎消失。N掺杂也对F2g峰的位置和强度也有明显的影响,如图4(b)所示。这说明Fe、N的掺杂改变了CeO2晶格中Ce-O键的键能和原子间距,导致电子云发生了迁移。同时,Fe、N的掺杂也对晶格的极化率发生变化。另外,Raman图谱在550~600 cm-1之间还存在一个比较宽的峰,此峰的形成反应了晶体中的氧空位浓度的变化[22-23]。
2.5 紫外-可见光谱分析
图5(a,b)分别为不同物质的量分数的Fe掺杂CeO2和不同氮源N-10%Fe-CeO2的紫外-可见光漫反射光谱图。可见纯CeO2的吸收带边在400 nm附近垂直陡峭,而掺杂Fe以后,吸收带发生明显红移,吸收带边扩展到可见光波段,缓慢降低。N的掺杂对10%Fe-CeO2有不同程度的影响,以浓氨水做氮源时扩展最大。利用半导体材料带隙计算公式:

图4 (a)Fe-CeO2与(b)N-10%Fe-CeO2的Raman图谱Fig.4 Raman spectra of(a)Fe-CeO2and(b)N-10%Fe-CeO2
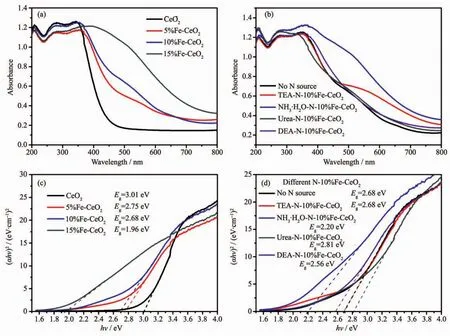
图5 不同Fe含量的Fe-CeO2(a)与不同氮源N-10%Fe-CeO2(b)的UV-Vis吸收图及(αhν)2~h关系图(c,d)Fig.5 UV-visible spectroscopy of Fe-CeO2(a)and N-10%Fe-CeO2(b)and their(αhν)2~h plots(c,d)

式中,α为吸光系数;h为普朗克常数;ν为光的频率;A为半导体材料的常数;Eg为半导体的禁带宽度;n为常数,通常材料带宽为直接型的n取1,间接型的n取4。氧化铈属于直接型带宽材料,因此n取 1。作(αhν)2~hν关系曲线,如图 5(c,d)所示。 曲线的切线与横坐标的截距即为该材料的带隙宽度Eg。从图中可见,随着掺杂Fe含量的增加,带隙宽度逐渐变小。据文献[24]报道,Fe掺杂使得导带宽度增加,导带底下移,并且在导带底附近形成了一个局域能级,在原来的禁带中也产生一个杂质能级,从而导致了禁带宽度变窄。而N的掺杂对10%Fe-CeO2的能带结构也有一定的微调作用,主要影响了其对可见光的吸收(图5(b))。浓氨水做N源时带隙宽度降低至2.20 eV,而尿素做氮源时,带隙宽度反而升高至2.81 eV。这说明不同的氮源掺杂对晶体的电子结构起到非常重要的影响。N掺杂进入CeO2晶格中,取代了O的位置。N比O更容易失去电子,因此降低了导带能级的高度。N的2p电子层与周围O的2p电子层形成杂化,在价带顶部产生新的杂质电子轨道,缩小了禁带宽度,从而促进了光生电子与空穴的产生。
2.6 光催化性能分析
图6为亚甲基蓝溶液在氙灯的照射下的光催化降解浓度变化曲线。可见在Fe-CeO2样品中,以10%Fe-CeO2的样品催化效率最高,掺杂Fe含量增加到15%后,催化速度反而有所减弱。但最终Fe-CeO2的降解率都在95%左右。而纯CeO2在同样条件下对亚甲蓝的降解率只有67%。掺杂N以后,10%Fe-CeO2的催化速率和催化效果产生了不同的变化。以浓氨水和二乙醇胺做氮源时,催化速度进一步提高,最终降解率达到97%。而尿素做氮源时反而使得其催化效果有所降低。这显然与N掺杂后,其禁带宽度的改变密切相关。禁带宽度越窄,可吸收的紫外线和可见光越多,产生的光生电子-空穴对越多,催化降解亚甲基蓝速度越快,最终降解率也越高。由于Fe3+具有未填满的d层电子轨道,容易捕获光电子成为Fe2+,也可捕获空穴成为Fe4+。因此当Fe掺杂含量较低时,可成为光生电子或空穴的陷阱,促进光生载流子的分离,从而起到提高催化性能的作用。然而当Fe掺杂含量过高,Fe可能会成为光生电子与空穴的复合中心,虽然禁带宽度变得更小,但其催化速率反而会降低[18-19]。氮掺杂后,N2p轨道与O2p轨道在导带底部形成杂化轨道,产生杂质能级,降低了导带能级高度,同时也在价带顶部形成局部能级[17]。但由于掺杂的氮源不同,N的掺杂含量和掺杂状态不同导致活性点数量和活性点的催化能力不一样,导致其催化性能也有所不同。掺杂Fe和N对CeO2电子结构的改变和相应的催化机理可用图7表示。不同的掺杂效应对10%Fe-CeO2的能带结构产生了不同的影响,从而影响了其最终的催化效果。Fe与N掺杂逐渐降低了导带的高度,并且在导带底或价带顶部形成局域能级,或在带隙中形成杂质能级,从而促进了光生电荷的产生。

图6 CeO2和不同含量Fe掺杂的Fe-CeO2(a)及不同氮源N-10%Fe-CeO2(b)作催化剂的光催化降解亚甲基蓝溶液性能Fig.6 Photocatalytic degradation of methylene blue solution with pure CeO2and Fe-CeO2as catalystic agents(a)and different nitrogensource N-10%Fe-CeO2as catalytic agents(b)
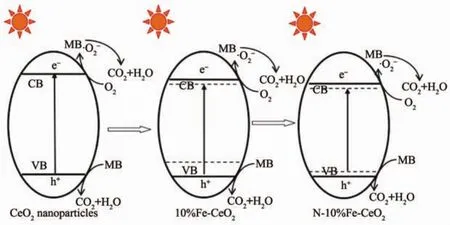
图 7 CeO2、10%Fe-CeO2和 N-10%Fe-CeO2的催化机理示意图Fig.7 Schematic diagram of catalytic mechanism of CeO2,10%Fe-CeO2and N-10%Fe-CeO2
2.7 光催化剂的循环使用性能
图8 为NH3·H2O-N-10%Fe-CeO2的循环使用的降解率结果(每次光催化时间为210 min)。降解率随着使用次数的增加,略有降低。经过5次使用后,光催化剂在210 min内对亚甲基蓝的降解率仍有89%,表明催化剂的光催化性能具有良好的稳定性。图8(b)对比了5次循环使用后的催化剂与制备态的催化剂的XRD图,并没有发现有明显变化。说明催化剂在使用前后结构没有改变,也几乎没有吸附其它物质。 因此,NH3·H2O-N-10%Fe-CeO2的光催化性能总体上比较稳定。在循环使用过程中,降解率有所下降的原因可能是催化剂在回收再利用过程中发生了微量的质量损失。
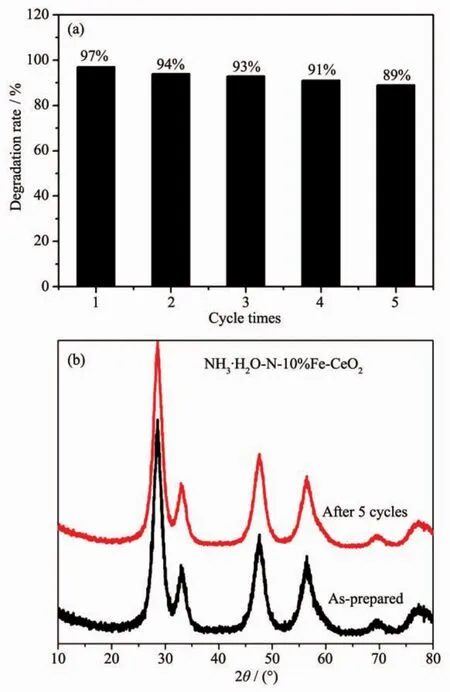
图8 (a)NH3·H2O-N-10%Fe-CeO2催化剂对亚甲基蓝溶液的降解率与循环使用次数的关系;(b)循环使用前后的XRD对比图Fig.8 (a)Relationship of photocatalytic degradation rate of methylene blue solution and use time with NH3·H2O-N-10%Fe-CeO2as catalytic agent;(b)XRD patterns comparison for as-prepared and after 5 cycles photocatalysis
3 结 论
本文采用溶剂热法合成了不同Fe掺杂含量的Fe-CeO2纳米粉体及不同氮源掺杂的N-10%Fe-CeO2纳米粉体,并对其光催化性能进行了研究。可得到以下结论:
(1)Fe掺杂引起晶格畸变,形成反应活性中心,产生杂质能级,降低禁带宽度。10%Fe-CeO2的催化速率最高,将纯CeO2对亚甲基蓝的降解率从67%提高到95%。
(2)掺杂过量Fe时,Fe会成为光生电子与空穴的复合中心,从而降低光生载荷的量子效率,降低其催化活性。
(3)不同氮源掺杂对10%Fe-CeO2的生长形貌具有重要影响,其中以浓氨水为氮源掺杂形成中空薄壁状N-10%Fe-CeO2粉体的禁带宽度最小,对亚甲蓝溶液的降解效率可提高到97%。
(4)以浓氨水为氮源制备的N-10%Fe-CeO2催化剂具有较好的性能稳定性。经5次循环使用,对亚甲基蓝溶液的光催化降解率仍高达89%。
参考文献:
[1]SU Lin-Feng(苏琳峰),GONG Jin-Feng(巩金峰),MENG Fan-Ming(孟 凡 明).Chinese Journal of Synthetic Chemistry(合成化学研究),2016,4(4):29-37
[2]QI En-Lei(齐恩磊),MAN Li-Ying(满丽莹),WANG Sun-Hao(王孙昊),et al.Chinese Journal of Materials Research(材料研究学报),2011,25(2):219-224
[3]ZHAO Xiao-Bing(赵晓兵),YOU Jing(游静),LU Xiao-Wang(陆晓旺),et al.J.Inorg.Mater.(无机材料学报),2011,26(2):159-164
[4]WU Jun-Ming(吴俊明),WANG Ya-Ping(王亚平),YANG Han-Pei(杨汉培),et al.Chinese J.Inorg.Chem.(无机化学学报),2010,26(2):203-210
[5]Liyange A D,Perera S D,Tan K,et al.ACS Catal.,2014,4(2):577-584
[6]WANG Jing-Xin(王敬欣).J.Chin.Rare Earth Soc.(中国稀土学报),2007,25(S1):82-88
[7]Wang W,Zhu Q,Dai Q G,et al.Chem.Eng.J.,2017,307:1037-1046
[8]YUAN Qiang(袁强),JIANG Ling(江玲),LI Hui(李辉),et al.Journal of Xiamen University:Natural Sciences Edition(厦门大学学报:自然科学版),2011,50(1):70-75
[9]Arul N S,Mangalaraj D,Chen P,et al.Mater.Lett.,2011,65:3320-3322
[10]Arul N S,Mangalaraj D,Han J.Mater.Lett.,2015,145:189-192
[11]Xie S L,Wang Z L,Cheng F L,et al.Nano Energy,2017,34:313-337
[12]ZHANG Guo-Fang(张国芳),ZHANG Yang-Huan(张羊换),GE Qi-Lu(葛启录),et al.Spectrosc.Spectr.Anal.(光谱学与光谱分析),2011,31(12):3315-3318
[13]LI Chang-Quan(李长全),LUO Lai-Tao(罗来涛),XIONG Guang-Wei(熊光伟).Acta Chim.Sin.(化学学报),2010,68(10):1023-1026
[14]GU Ming-Jie(顾明杰),LI Rui-Xing(李锐星),SONG Xiao-Zhen(宋晓贞).Journal of Ceramics(陶瓷学报),2013,34(2):135-138
[15]Mao C J,Zhao Y X,Qiu X F,et al.Phys.Chem.Chem.Phys.,2009,40(3):5633-5638
[16]Wu C L.Mater.Lett.,2015,139:382-384
[17]Ren R K,Zhang M J,Meng J,et al.Chin.Phys.B,2017,26(3):036102
[18]HUANG Dong-Sheng(黄东生),CHEN Chao-Feng(陈朝凤),LI Yu-Hua(李玉花),et al.Chinese J.Inorg.Chem.(无机化学学报),2007,23(4):738-742
[19]JIAN Li-Xin(翦立新),YIN Xiao-Qiu(殷小秋),XIANG Jian-Nan(向建南),et al.Journal of Hunan University:Natural Sciences Edition(湖南大学学报:自然科学版),2006,33(1):79-82
[20]Sun C W,Li H,Chen L Q.Energy Environ.Sci.,2012,5:8475-8505
[21]Toru Y,Peter H.Appl.Surf.Sci.,2008,254:2441-2449
[22]Jeyanthi C E,Siddheswaran R,Kumar P,et al.Ceram.Int.,2014,40(6):8599-8605
[23]Xu B,Zhang Q T,Yuan S S,et al.Catal.Today,2017,281(1):135-143
[24]Tian D,Zeng C H,Fu Y C,et al.Solid State Commun.,2016,231/232:68-79
猜你喜欢
中国粉体技术(2022年5期)2022-09-06
中国粉体技术(2022年2期)2022-03-19
当代作家(2021年11期)2021-12-17
粉末冶金技术(2021年1期)2021-03-29
粉末冶金技术(2021年1期)2021-03-29
科学(2020年4期)2020-11-26
科学(2020年4期)2020-01-11
数学物理学报(2019年5期)2019-11-29
中国酿造(2016年12期)2016-03-01
中国酿造(2014年9期)2014-03-11
