
碳纳米管基非贵金属催化剂在电催化氧化还原中的应用研究进展
2018-05-05 06:22王启晨雷永鹏陈志彦罗世彬
无机化学学报 2018年5期
王启晨 王 璟 雷永鹏 陈志彦 宋 垚 罗世彬
(1中南大学,航空航天学院&轻质高强结构材料重点实验室,长沙 410083)
(2国防科技大学理学院,长沙 410073)
(3武汉科技大学省部共建耐火材料与冶金国家重点实验室,武汉 430081)
(4国防科技大学基础教育学院,长沙 410073)
(5高性能陶瓷和超微结构国家重点实验室,上海 200050)
(6中南林业科技大学材料科学与工程学院,长沙 410004)
0 引 言
面对日益严重的能源危机和环境污染等一系列问题,迫切需要发展新型清洁、高效、安全的能源,这已成为全世界范围的共识。燃料电池和金属-空气电池具有能量转化效率高、绿色环保等优点,在解决能源和环境问题方面具有非常大的潜力,可广泛应用于航空航天、载运工具和固定电站等[1-3]。以氢-氧燃料电池为例,其工作原理是氢气由阳极进入,在阳极发生电化学氧化反应,产生成对的电子和质子。质子经质子交换膜,由阳极迁移到阴极,而电子经外电路转移到达阴极。氧气由阴极进入,结合外部的电子发生氧还原反应(oxygen reduction reaction,ORR)。电子在迁移过程中经外电路做功形成回路,产生电流,化学能转化为电能[4-5]。ORR存在动力学过程缓慢、过电势高等问题,是限制燃料电池性能提升的瓶颈[6-7]。一般来说,ORR可通过直接四电子过程或间接2步两电子过程进行。理想的ORR应该通过四电子过程进行,因为两电子过程会形成有害的过氧根离子,降低催化活性[8]。目前贵金属铂(Pt)及其合金是典型的ORR电催化剂,但Pt储量少、价格高且易中毒失活,这样一来,开发低成本、高性能的非贵金属ORR催化剂就成为实现燃料电池和金属-空气电池大规模应用的关键[9-10]。碳材料来源丰富,有很好的耐腐蚀性和化学稳定性[11],通过对碳材料的电子性质、化学组成和微观结构的精细调控,可使其电催化活性与贵金属催化剂相媲美。因此,碳材料受到相关研究人员的广泛关注。
碳纳米管(CNTs)具有本征sp2杂化结构、优异的导电性、高的比表面积、良好的化学稳定性等突出优点,在电极材料、催化剂载体、超级电容器等方面展示出广阔的应用潜力[12-15]。目前,碳纳米管的制备方法主要包括电弧法[16]、离子或激光束法[17]、化学气相沉积(CVD)[18],催化裂解[19-20]等。 有报道[21]称多壁碳纳米管具有一定的电化学活性,但与壁层数密切相关,2~3层碳纳米管的活性最优。另一方面,碳纳米管表面具有π电子高度离域化的类石墨层结构,碳纳米管之间存在很强的范德华力,极易发生团聚,限制了其在电催化ORR中的应用。总的来说,碳纳米管作为一类电中性的碳材料,化学惰性大且溶解性较低。因此,必须对其表面进行功能化处理。用浓硫酸、浓硝酸等强氧化性酸对碳纳米管进行浸渍处理或者利用等离子体气体选择性氧化刻蚀,可以提高其分散性并改善与其他材料的复合行为[22-25]。Liu等[26]设计出一种咪唑配位卟啉修饰的多壁碳纳米管,半波电势高达0.922 V,超过商业Pt/C催化剂47 mV。
作为ORR催化剂需具备以下基本条件:(1)良好的导电性;(2)高的比表面积;(3)丰富的活性位。因此,主要有以下途径对碳纳米管改性:非金属掺杂、过渡金属-氮-碳纳米管、负载过渡金属及其衍生物(氧化物、碳化物、氮化物、硫化物等)、负载单原子、与其他碳材料(石墨烯、多孔碳、碳纳米纤维)复合等,本文对上述各种催化剂的合成、性能及在电池中的应用等进行了综述。此外,还简要介绍了碳纳米管基自支撑电极的研究进展。最后,对碳纳米管基ORR电催化剂的研究做了展望。
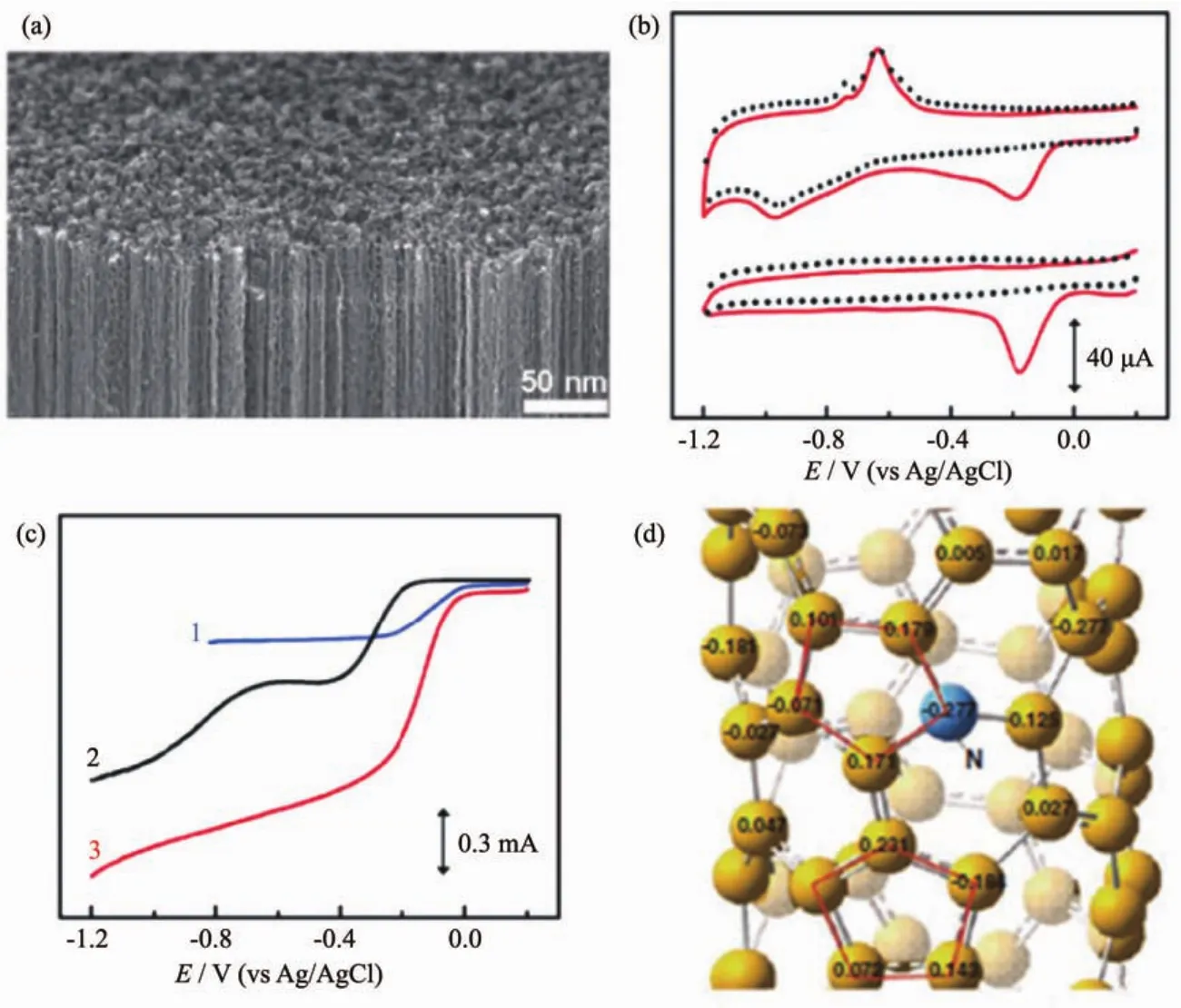
图1 (a)VA-NCNTs的SEM图;(b)VA-NCNT/GC的CV曲线;(c)不同样品的LSV曲线(曲线1,2和3分别代表着NA-CCNT/GC,Pt-C/GC和NA-NCNT/GC电极);(d)NCNTs的电荷密度分布图[30]Fig.1 (a)SEM image of VA-NCNTs;(b)CV curves for ORR at the unpurified(upper)and electrochemically purified(bottom)VA-NCNT/GC electrodes in the argon-protected(dotted curves)or air-saturated 0.1 mol·L-1KOH(solid red curves)at the scan rate of 100 mV·s-1;(c)LSVs curves at the Pt-C/GC(curve 1),VA-CCNT/GC(curve 2),and VA-NCNT(curve 3)electrodes;(d)Calculated charge density distribution for the NCNTs[30]
1 非金属掺杂碳纳米管
1.1 氮掺杂
对碳纳米管掺杂异质元素一方面可以优化材料表面电子结构和表面电荷分布,增强对O2的吸附能力[27-28],还可以引入更多的缺陷(边缘、空位等),促进ORR催化[29]。含氮前驱体来源广泛、组成结构形貌可控,因此N掺杂碳纳米管(N-CNTs)受到广泛关注。如图1a所示,Dai等[30]制备了富电子N掺杂的sp2杂化垂直碳纳米管阵列(VA-NCNTs),碱性条件下ORR活性接近Pt/C(图1(b,c))。由于N原子电负性较C原子大 (N电负性为3.04;C电负性为2.55),理论计算表明N掺杂使邻近C原子带正电荷,有利于吸附O2从而促进ORR进行(图1d)。N的键合形式和含量对催化性能具有至关重要的影响[31-33]。一般而言,掺N碳材料中,N有3种键合结构(图 2),分别为吡啶氮(398.6 eV)、吡咯氮(400.6 eV)和石墨氮(401.6 eV)[34]。Ishikawa等[35]制备了一系列N掺杂的垂直碳纳米管,发现其ORR活性与吡啶氮正相关。掺杂量的多少并不是唯一影响N-CNTs的因素,实现N原子对目标位置精确掺杂是提高活性的首要条件。Wei等[36]通过CVD法制备了CNT为核、N-CNTs为壳的复合催化剂(CNT@N-CNT),N元素主要分布在外层壳内,这种富N的壳层更易于与O2充分接触,大大提高了活性位点的利用率,表现出与Pt/C相近的ORR活性。Zhang等[37]利用水热法结合热解制备出一种N-CNTs气凝胶。受益于高比表面积(869 m2·g-1)、高导电性(10.9 S·m-1)以及气凝胶自身的多孔网络结构,该材料表现出良好的催化活性和稳定性。该工作为制备高比表面的NCNTs提供了一种新思路。
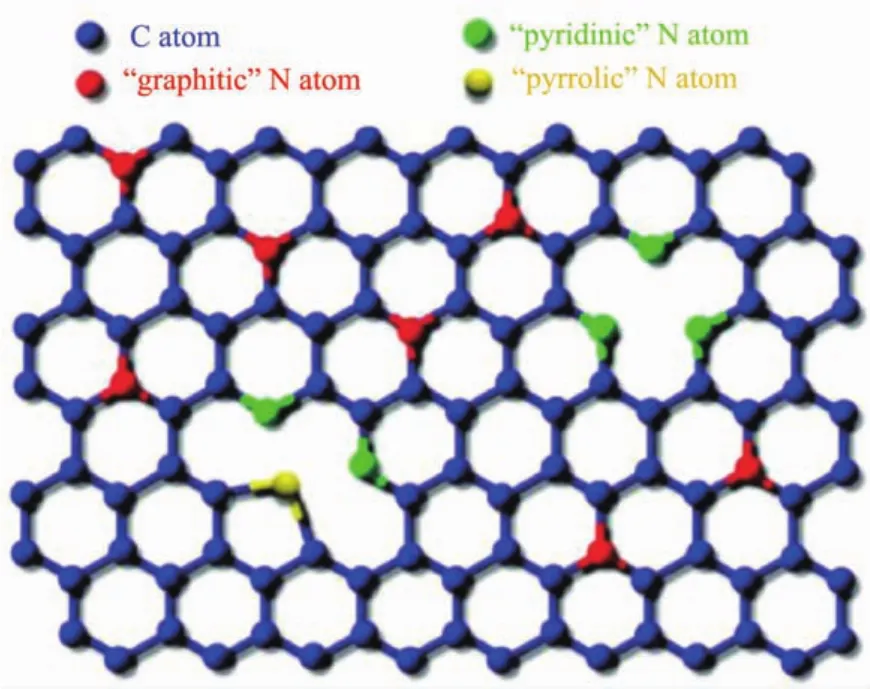
图2 氮在石墨结构中的掺入位置[34]Fig.2 Schematic representation of N bonding configurations[34]
除掺N元素外,在碳纳米管中掺入S,B,P等元素也可以改变电子结构和化学性质,从而调控催化活性[38-39]。Hu等[40]制备的B掺杂碳纳米管(BCNTs)表现出较高的ORR活性。与N原子不同,B本身缺电子。理论计算表明,引入杂原子提高sp2碳材料ORR活性的关键是活化了π电子,促进O2的利用,而不在于掺杂原子本身是富电子还是缺电子。Li等[41]一步热解制备了S掺杂碳纳米管(SCNTs),起始电势为-0.082 V(vs Ag/AgCl),在-0.40 V时对应的电流密度为5.35 mA·cm-2,同时还表现出优异的稳定性和抗甲醇毒化能力。
1.2 多组分掺杂碳纳米管
在引入N元素的同时,引入B,S,P等元素进行双掺杂甚至多元素掺杂能进一步提升ORR活性,这主要是因为多组分掺杂提供了更多的活性位点,适当的掺杂比例能够增强协同效应[42-44]。Wang等[45]研究了B,N共掺杂碳纳米管(VA-BNCNT)的ORR性能。相比单掺杂,双掺杂产物的起始电势和极限扩散电流密度都得到显著提高,活性与Pt/C相当,证实了双元素掺杂的协同效应。如图3a所示,Su等[46]选用不同的硫源与N-CNTs经水热和热处理,制备了S,N共掺杂的竹节状碳纳米管(SN-CNTs),S和N元素均匀分散在碳纳米管表面(图3b)。在碱性电解液中,SN-CNTs的ORR峰电势和峰电流都要优于N掺杂或未掺杂的CNTs。酸性条件下的ORR性能比N-CNTs也有很大提高。这主要是由于S元素的引入改变了N的价态,造成轨道和电荷密度的不对称,促进了O2吸附。Woo等[47]制备了B,P,N共掺杂的竹节状碳纳米管(BPNDC),竹节状构型有利于暴露更多的石墨化边缘,增加ORR的活性位。同时,B,P,N三元素掺杂有效提升了ORR性能,优于双掺杂或单掺杂样品。
此外,利用极性分子修饰碳纳米管也是一种提高ORR活性的有效策略。季铵型阳离子聚合物(PDDA)具有很强的吸电子能力,可通过静电吸附在带负电的酸氧化后的碳纳米管表面。Dai等[48]通过简单的旋转涂布和过滤制备出PDDA杂化的垂直碳纳米管阵列 (PDDA-ACNT),发现修饰后的样品ORR活性明显提高了。他们认为PDDA具有很强的吸电子能力,通过分子内的电荷转移使相邻的C原子带正电,从而造成碳纳米管表面电子的离域,增强其催化ORR的能力。
杂原子掺杂的碳纳米管ORR催化剂活性较高、成本低、寿命长,然而目前这类催化剂在碱性条件下催化活性较高,但在酸性条件下的活性并不高。主要原因可归结于以下3点:(1)酸性介质比碱性介质具有更强的腐蚀性;(2)含氮的活性位点易被质子化,导致在催化ORR过程中失效[49];(3)异质元素掺杂多通过热处理实现,但高温下容易分解导致掺杂量低,限制了催化活性的提高。如何提高杂原子掺杂碳纳米管催化剂在酸性条件下的性能仍面临很大挑战。
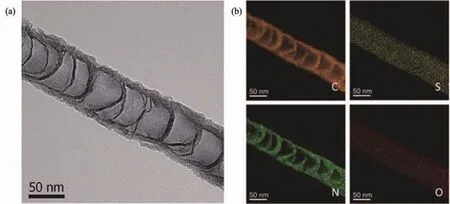
图3 (a)SN-CNTs的TEM图;(b)对应的C,S,N和O的mapping图[46]Fig.3 (a)TEM image of SN-CNTs;(b)Corresponding mappings of C,S,N and O[46]
2 过渡金属-氮-碳纳米管
过渡金属-氮-碳(M-Nx-C,M=Fe,Co)ORR 催化剂活性较高(特别是在酸性溶液中)、稳定性好、成本低、环境友好,是近年来非常热门的一类阴极催化剂,前驱体的选择和合成工艺的优化对催化性能至关重要[50]。尽管目前对金属是否参与活性位点的组成存在分歧,但过渡金属的确对提高ORR活性起到了积极作用。借助穆斯堡尔谱 (Mössbauer spectroscopy)和X射线吸收精细结构(EXAFS)等表征手段,结合理论计算,越来越多的研究认为该类催化剂中存在的M-Nx-C结构是ORR的活性位点[51-54]。
Yasuda等[55]利用π-π相互作用将酞菁铁吸附在碳纳米管阵列(图4a),通过高温热解在碳纳米管表面覆盖一层均匀的Fe-N共掺杂碳层(Fe-N-C/VACNT)。与多壁碳纳米管相比,以碳纳米管阵列为载体不但获得了更大的比表面积(504 m2·g-1),而且克服了多壁碳纳米管易团聚的不足,在阵列表面形成高密度的Fe-N活性位点(图4b)。该催化剂在酸性电解液中的起始电势和半波电势分别为0.97、0.79 V(图 4c)。
金属有机骨架化合物(MOFs)是一种多孔晶体材料,由金属离子或金属簇与有机配体联结而成。Ahn等[56]以多孔碲纳米管作为牺牲模板,合成Fe,N共掺杂的多孔碳纳米管(pCNT@Fe@GL)。制备过程中,金属有机骨架化合物(ZIF-8)首先在碲纳米管表面成核,然后再引入含Fe离子的超薄聚多巴胺层。热处理后,通过酸洗除掉Te和Zn,获得含有Fe-Nx的多孔碳骨架锚定在碳纳米管表面。该催化剂比表面积高达1 380 cm2·g-1,且Fe-Nx活性位均匀的分散到碳骨架中,在碱性和酸性介质中的半波电势分别为0.867、0.818 V。Zhang等[57]将碳纳米管引入铁锌双金属Zn-ZIF-8,热处理过程中Zn气化产生了更多的微孔,有利于FeNx活性位的分散,碳纳米管提高了导电性与传质效率,再浸入糠醇再热解,获得了更多的活性位点。将最终得到的催化剂应用于质子交换膜燃料电池,功率密度高达820 mW·cm-2。Yang等[58]发展了一种Fe,N共掺杂的碳纳米管海绵作为高效的酸性ORR催化剂。Liu等[59]通过原位固相热解制备了一种超薄、褶皱状的Co,N共掺杂碳纳米管ORR催化剂。

图4 (a)Fe-N-C/VA-CNT的合成示意图;(b)Fe-N-C/VA-CNT的TEM和HRTEM图;(c)不同样品在O2饱和的0.5 mol·L-1H2SO4中的LSV曲线[55]Fig.4 (a)Schematic diagram of the synthesis of Fe-N-C/VA-CNT;(b)TEM and HRTEM images of Fe-N-C/VA-CNT;(c)LSV curves of different catalysts in O2-saturated 0.5 mol·L-1H2SO4solution[55]
3 碳纳米管负载过渡金属及其衍生物
负载型金属催化剂在整个工业催化领域发挥着十分重要的作用。在催化过程中,纳米颗粒容易出现溶解或团聚现象,进而导致催化剂失活[60]。将无机纳米颗粒(过渡金属、金属氧化物、碳化物、氮化物和硫化物等)负载到改性的碳纳米管上,使其不仅可以作为导电载体,也可与电化学活性高的金属颗粒及其衍生物发生耦合作用,优化催化剂表面的电子结构,提高活性和稳定性[61-64],因而成为非贵金属ORR催化剂的研究热点之一。
3.1 金属/碳纳米管
Bao等[65]制备了豆荚状氮掺杂纳米管包裹Fe纳米颗粒的催化剂(Pod-Fe),尽管Fe不与电解液直接接触,但Fe向周围碳层转移电子,降低了碳纳米管表面的局部功函数,从而形成有利于ORR的活性中心。Wang等[66]以三聚氰胺为氮源,制备了长径比均一的含Co的N掺杂竹节状碳纳米管(Co-NBCNT),其在碱性条件下的半波电势和扩散电流都优于商业Pt/C。同时发现,包覆在碳纳米管内的Co颗粒对ORR活性起着至关重要的作用,Co活化了周围的石墨化碳层,使碳层具有ORR活性。MOFs本身的有机配体可形成碳框架结构,并且有机配体中通常含有杂原子,可在一定温度下热处理得到杂原子掺杂的碳材料。因此,近年来基于MOFs制备纳米多孔碳材料备受关注[67-69]。最近,Mai课题组[70]设计了一种在430℃即可定向生长碳纳米管的方法。热处理过程中产生的还原性气体首先将Co2+转变为Co颗粒,进而催化残余的有机配体生成碳纳米管(图5a)。如图5b所示,650℃得到的样品具有最好的ORR活性,半波电势为0.85 V,且在0.20~0.75 V电势区间内有明显的电流平台,表明催化ORR主要是4e-过程。结合紫外光电子能谱(UPS)和 DFT(密度泛函理论)计算(图 5(c,d))可知,石墨-N和碳纳米管限域的Co之间的协同效应增加了费米能级附近的态密度,降低了功函数,从而提升了ORR性能。因此,MOFs是一种合成碳纳米管基ORR催化剂的有效前驱体。Xia等[71]报道了一种从ZIF-67衍生的N掺杂碳纳米管空心框架结构,Co颗粒被碳纳米管包覆提供了大量的活性位,碱性条件下的ORR活性和稳定性都优于商业Pt/C。

图5 (a)N-CNTs-650的SEM图,插图为对应的HRTEM图;(b)不同催化剂在O2饱和的0.1 mol·L-1KOH中的LSV曲线;(c)紫外光电子能谱图,插图为放大图;(d)Co限域在N-CNT的催化ORR示意图[70]Fig.5 (a)SEM image of N-CNTs-650,the inset shows the corresponding HTTEM image;(b)LSV curves of different catalysts in O2-saturated 0.1 mol·L-1KOH solution;(c)UPS spectra collected using He I(21.2 eV)radiation.Inset shows the enlarged view;(d)Schematic of Co confined in N-CNTs for the ORR process[70]
ORR是典型的耗气反应,氧气在电解液中的溶解度低,氧气浓度不足就会降低催化剂的活性[72]。针对这一问题,Sun等[73]通过在碳纳米管阵列表面修饰聚四氟乙烯,构筑微纳结构“超亲气”电极,使气体能够通过扩散层快速到达电极表面从而提高ORR效率。经修饰后的催化剂性能大幅提升,甚至在较大的过电势区域内优于Pt/C。该设计思路对于其他耗气反应也具有普遍的指导意义。
3.2 金属氧化物/碳纳米管
金属氧化物是一类重要的ORR催化剂,但低的导电性和比表面积限制了其本征催化性能的发挥[74]。将金属氧化物负载到改性的碳纳米管上,可以有效地解决上述问题。目前,钴、铁和锰基氧化物在碱性溶液中表现出很好的ORR活性[75-78]。如图6(a~c)所示,Dai等[79]通过溶剂热和氨气处理制备了尺寸均一的CoO并负载于N掺杂的CNT(CoO/N-CNT)。以其为空气电极组装成锌-空电池,最大功率密度为265 mW·cm-2(图6d),高于同等条件下的Pt/C性能(250 mW·cm-2)。 此外,在 5、50 mA·cm-2下连续放电,放电平台没有明显压降(图6e)。Seo等[80]将不同尺寸的 CoOx颗粒(4.3,6.3,7.5 和 9.5 nm)负载到碳纳米管,发现CoOx的尺寸对催化剂ORR活性影响不大,原位电化学X射线吸收谱结果表明催化过程中CoOx主要以Co3O4和CoOOH存在。Yang等[81]制备出多种金属氧化物(MnO2,ZnO,NiO,Fe3O4,TiO2)修饰的3D垂直碳纳米管阵列(ACNTs),其中MnO2-ACNTs在碱性电解液中表现出优异的ORR活性、稳定性和抗甲醇毒化能力。
尖晶石型氧化物(AB2X4)价态可调、来源丰富且价格低廉,在碱性介质中表现出优异的ORR性能,日益受到关注[82-86]。Yuvaraj等[87]在N掺杂多壁碳纳米管上负载了立方尖晶石结构的MnCo2O4微球,N掺杂和金属氧化物的协同效应显著提升了ORR性能。 Li等[88]通过水热法将 MFe2O4(M=Co,Ni)纳米颗粒负载到多壁碳纳米管(MFe2O4/CNT),并用于锂-氧电池,电流密度为 200 mA·g-1条件下,首圈放电容量为3 670 mAh·g-1。他们认为该性能主要归结于高分散的CoFe2O4颗粒作为反应活性位和碳纳米管优异的导电能力。尖晶石氧化物的催化性能和尺寸密切相关,Shi等[89]通过调控反应温度制备了不同尺寸(2.0、3.9和5.4 nm)的CoMn2O4量子点/碳纳米管复合物(CoMn2O4/CNT)。适宜的尺寸能够调控能带结构和提高表面吸附O2能力,进而提高体系中载流子的浓度。CoMn2O4为3.9 nm时催化剂半波电势为0.89 V。Liu等[90]通过水热法合成了ZnCo2O4量子点负载的N-CNTs,以其为空气电极用于锌-空电池,比容量和能量密度分别为 428.47 mAh·g-1和595.57 Wh·kg-1, 接近商业Pt/C性能 (548.35 mAh·g-1;762.21 Wh·kg-1)。

图6 (a)CoO/N-CNT的SEM图和(b)HRTEM图;(c)CoO/N-CNT示意图;(d)电池的放电极化曲线和对应的功率密度曲线;(e)锌-空电池在5和50 mA·cm-2下的放电极化曲线[79]Fig.6 (a)SEM image and(b)HRTEM image of CoO/N-CNT;(c)Schematic diagram of CoO/N-CNT;(d)Discharging polarization curves and the corresponding power curves;(e)Discharging polarization curves at 5 and 50 mA·cm-2 in Zn-air batteries[79]
3.3 金属碳化物/碳纳米管
过渡金属碳化物具有较高的稳定性和抗腐蚀能力[91-92],其中Fe3C是目前研究最为广泛的过渡金属碳化物ORR催化剂。如图7(a,b)所示,Yang等[93]基于软模板F127和热处理将Fe3C包覆在竹节状碳纳米管(BCNFNHs),软模板和Fe(NO3)3的加入对样品的形貌至关重要。在0.1 mol·L-1KOH溶液中,半波电势为0.861 V(图7c),塔菲尔斜率为91.2 mV·dec-1。在 0.5 mol·L-1H2SO4溶液中,起始电势是0.886 V(图 7d), 塔菲尔斜率是 108.6 mV·dec-1。3 000次加速CV循环测试后,ORR活性几乎保持不变。Wei等[94]在碳纳米管表面负载Fe3O4纳米颗粒,随后包覆一层聚多巴胺,通过热处理将聚多巴胺层转化为多孔氮掺杂碳层,Fe3O4原位还原为FexC纳米颗粒。碳纳米管的高导电性有利于电子的高效传输,表面碳层的多孔结构进一步增大了该催化剂的比表面积(245 m2·g-1)。此外,FexC与氮掺杂碳层之间的强化学耦合改善了起始电势和电流密度。Guan等[95]将基于FeZn的ZIFs作为前驱体进行限域热解,将Fe3C纳米晶负载在“海胆”状多孔氮掺杂碳纳米管-碳多面体上,并用于ORR催化。Jiang等[96]发展了一种由多孔碳层包覆的碳纳米管,同时含有碳层包覆的Fe/Fe3C(Fe@C),该催化剂在0.1 mol·L-1KOH中半波电势为0.899 V。结合电子能量损失谱 (EELS)和X射线吸收精细结构谱(EXAFS)分析,证实在邻近碳层包覆的Fe@C周围存在Fe-Nx活性位点,且在含有Fe@C时,催化剂活性与Fe-Nx活性位点的数量正相关。DFT理论计算也表明当Fe-Nx活性位点附近含有金属铁原子时,Fe-Nx活性位的最高占据轨道升高,与氧分子的p轨道的更易重叠,利于O2吸附,从而加速了ORR过程。此外,Pang等[97]利用微波辅助的方法制备出直径3~6 nm球形Mo2C纳米颗粒,均匀负载到碳纳米管上,作为ORR催化剂。
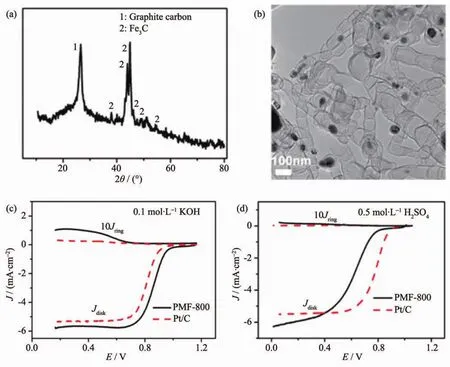
图7 PMF-800的(a)XRD图和(b)TEM图;PMF-800和Pt/C催化剂在O2饱和的0.1 mol·L-1KOH(c)和(d)0.5 mol·L-1H2SO4中的 LSV 曲线[93]Fig.7 (a)XRD patterns and(b)TEM image of PMF-800;LSV curves of PMF-800 and Pt/C catalysts in O2-saturated 0.1 mol·L-1KOH(c)and(d)0.5 mol·L-1H2SO4solution[93]
3.4 金属氮化物(硫化物)/碳纳米管
除此之外,金属氮化物也是重要的酸性ORR电催化剂。N原子掺杂到金属晶格间中显著改善了过渡金属的d带电子结构,在费米能级面有更高的电子密度,增强了O2接受电子的能力[98]。CuN[99]、TiN[100]、MoN[101]与碳纳米管的复合物都表现出优异的活性。金属硫化物是一种廉价的材料,也可作为ORR催化剂[102],目前报道的主要有Co9S8[103]和Cu9S8[104]等。Han等[105]利用共沉淀再硫化的方法在N掺杂碳纳米管表面均匀生长出15 nm的NiCo2S4(NiCo2S4/N-CNTs),ORR半波电势为0.78 V,连续工作25 h后电流保持率为88.5%。组装成的锌-空气电池在0.77 V时的最大功率密度达到147 mW·cm-2,接近Pt/C电极 (181 mW·cm-2)。分析其优异ORR活性的原因,主要有:(1)小尺寸且均匀分散的NiCo2S4纳米颗粒提供更多的活性位点;(2)NiCo2S4与N掺杂CNT强耦合作用有利于高效的电子转移;(3)协同效应优化电子结构,促进O2吸附和活化。
4 碳纳米管负载单原子
金属-载体相互作用是负载型金属催化剂的基本结构特征,也是影响催化性能的关键因素,一般认为金属颗粒尺寸越小越好。单原子催化剂是负载型催化剂的极限状态,实现了金属原子100%的利用率[106-107]。众所周知,最大可能暴露/提高催化剂活性位点的数量/密度,可以最大幅度地提升性能。单原子催化剂中的金属以单原子的形式均匀分布在载体上作为活性中心,实现了催化反应的高活性和高选择性,同时也为催化机理的研究提供了新的解决方案[108]。
Lin等[109]以碲纳米线为模板,含氮小分子和无机盐作为氮源,一步水热结合热处理制备出负载Fe单原子的碳纳米管气凝胶(Fe-N-CNTAs)。Fe含量仅为0.09%(n/n),但ORR活性优于Pt/C,这来源于活性中心的不饱和配位状态以及与载体间稳定的相互作用。如图8a所示,Chen等[110]制备了单分散Fe-Nx活性位锚定的N,S共掺杂碳纳米管-石墨烯复合网络(S,N-Fe/N/C-CNT)。高角环形暗场扫描透射电子显微镜(HAADF-STEM)证实了Fe以高分散、单原子的形式存在 (图8b),EXAFS结果表明Fe只与N配位(图8c)。受益于高密度Fe-Nx活性中心、分级孔结构和优异的电子传输能力,催化剂的半波电势为0.85 V,极限扩散电流密度为6.68 mA·cm-2(图8d)。将其作为空气电极的锌-空电池最大功率密度高达 102.7 mW·cm-2,超越 Pt/C 电极(95 mW·cm-2)。此外,石墨相氮化碳(g-C3N4)是一种典型的具有sp2杂化形成的π共轭体系的聚合物半导体,在光催化领域具有广泛应用[111-112]。相比N掺杂的碳纳米管,g-C3N4具有更特别的N键合形式和较大的N配位空间,可锚定金属单原子。Zheng等[113]在碳纳米管上制备出一系列g-C3N4配位的过渡金属(M-C3N4;M=Cr,Mn,Fe,Co,Ni,Cu,Zn)。理论计算发现 Fe,Co,Ni与g-C3N4配位后具有更强的O2结合能和结构稳定性。以Co为例,作者认为锚定在g-C3N4中的Co-N2单体是Co-C3N4/CNT催化剂的活性中心。在碱性条件下,与商业Pt/C的半波电势相当,且表现出优异的稳定性。Guo等[114]通过一步原位限域热解将Co原子锚定在N,P共掺杂的碳纳米管上,在酸性和碱性条件下均表现出优异的ORR活性。
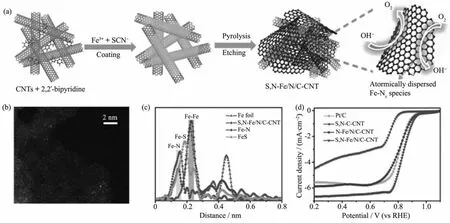
图8 (a)S,N-Fe/N/C-CNT合成示意图;(b)高角环形暗场扫描透射电子显微镜(HAADF-STEM)图;(c)扩展X射线吸收精细结构谱图;(d)不同催化剂在O2饱和的0.1 mol·L-1KOH中的LSV曲线[110]Fig.8 (a)Schematic diagram and(b)HAADF-STEM image of the synthesis of S,N-Fe/N/C-CNT;(c)EXAFS spectra;(d)LSV curves of different catalysts in O2-saturated 0.1 mol·L-1KOH solution[110]
5 碳纳米管-其它碳材料
碳纳米管在合成过程中容易发生团聚或者堆叠,导致其比表面积降低和活性位点减少,从而无法充分发挥其应有的性能。将碳纳米管与其他具有丰富活性位的碳材料(如石墨烯、多孔碳、碳纳米纤维等)复合形成C-C键并构筑三维(3D)网络结构,CC键强相互作用能使材料在催化过程中保持稳定,有效地增大比表面积,提高催化剂的导电性,大量的孔隙还可以促进反应物扩散并减小传质阻力。这种思路是基于1D、2D碳材料构建碳基3D电催化剂的有效方法,在 ORR[115]、析氢[116]、析氧[117]和 CO2还原[118]等各种电催化反应中都有很好的应用。
5.1 碳纳米管-石墨烯
Cheng等[119]在3D多孔石墨烯的缺陷处原位生长了N掺杂碳纳米管(N-CNTs-HGF),石墨烯网络具有很好的电子传输性能,同时多级孔结构促进离子快速传输,N-CNTs-HG表现出较高的起始电势(1.08 V)和低的塔菲尔斜率(72 mV·dec-1)。 Gao 等[120]将石墨烯和碳纳米管可控组装石墨烯/碳纳米管杂化纳米球,并通过N,P共掺杂引起碳原子电荷离域和电荷密度不对称,提高了活性位数量,在碱性体系中ORR性能优于Pt/C。Zhou等[121]将石墨烯量子点负载到多壁碳纳米管 (GQD-MWCNT),ORR催化主要以4e-路径进行,活性接近商业Pt/C。最近,我们课题组以氧化石墨烯为原料,通过热处理过程中Fe催化生长碳纳米管,原位构建了3D石墨烯/碳纳米管复合结构[122],包括Fe-Nx活性位、Fe@C包覆结构及N掺杂石墨烯与碳纳米管的复合物(NG/NCNTs),NG/NCNTs杂化结构有利于电子转移和传质。该催化剂在碱性条件下表现出优异的ORR活性,作为空气电极用于锌-空电池,其最大功率密度(101.2 mW·cm-2)与 Pt/C 电极(101.9 mW·cm-2)相当。

图9 (a)NG/Co-CNT/CNF的TEM图;(b)不同样品在O2饱和的0.1 mol·L-1KOH中的LSV曲线;(c)不同样品在0.44 V时的电子转移数比较;(d)NG/Co-CNT/CNF催化ORR的机理图[126]Fig.9 (a)TEM image of NG/Co-CNT/CNF;(b)LSVs(the dotted line is the LSV of NG/Co-CNT/CNF with a loading of 0.48 mg cm-2)at 1 600 r·s-1of different samples in 0.1 mol·L-1KOH;(c)Electron transfer numbers at 0.44 V of different samples;(d)Scheme of the 3D interconnected structure of NG/Co-CNT/CNF[126]
5.2 碳纳米管-多孔碳
Li等[123]以SiO2颗粒为模板,结合CVD和氨气热处理,制备出一种N掺杂碳纳米管网络的介孔碳(CNT/N-C)。介孔结构提供了有效的离子扩散通道,碳纳米管网络保证了电子的高效传输。在0.1 mol·L-1KOH中,CNT/N-C的起始电势和半波电势分别比商业Pt/C高59和30 mV,且抗甲醇毒化能力和耐久性测试都超过Pt/C。Wang等[124]以MOF-5和尿素作为前驱体,引入Ni元素提高石墨化程度,900℃处理得到N掺杂多孔碳和碳纳米管的复合物(NGPC/NCNT-900),表现出优秀的活性、稳定性和耐甲醇毒化能力,该催化剂是由MOFs转化制得的性能最好的ORR催化剂之一。Qiao等[125]结合软、硬模板法制备了分级有序多孔Fe,N共掺杂碳材料(Fe-N-CNT-OPC)。热解过程中,Fe催化原位生成碳纳米管并与多孔碳相互交错连成网络结构。该催化剂具有高活性的Fe-N活性位,分级孔有助于传质和暴露活性位,原位生成的碳纳米管则提高了材料的导电性。
本课题组结合静电纺丝和全固相热解法[126],在含Co碳纳米纤维上原位生长N掺杂石墨烯和N掺杂碳纳米管,得到3D网络结构(NG/Co-CNT/CNF)(图9a)。如图9(b,c)所示,该催化剂的起始电势达0.948 V,4e-过程催化ORR,稳定性和耐甲醇毒化能力高于商业Pt/C。优异的ORR性能主要是由于这种原位生长的3D网络结构有利于活性位点的暴露和反应物的传质以及电子转移(图9d),而被石墨碳包裹的Co颗粒也能提升ORR活性。Guo等[127]合成了N掺杂碳纳米管/碳纳米纤维杂化结构(NCNT/CNFs),实现了2种碳材料的原子尺度连接。该结构不仅为电子传输和离子扩散提供了3D连通网络,而且N掺杂显著地改变催化剂表面电子分布。
6 碳纳米管基自支撑电极
为更好地满足人们对柔性、可穿戴电子产品日益增长的需求,发展柔性电池迫在眉睫[128-129]。目前,柔性金属-空气电池主要是将空气电极、电解质膜和负极叠加组成的三明治结构,粉体催化剂需要和高分子粘结剂 (如Nafion)及金属集流体等共同使用,这会增加电极材料同基底的附着力及界面接触电阻,进而降低电池的能量效率(如功率密度、比容量和循环稳定性等)[130-132]。发展高性能的自支撑电极是解决上述问题的关键。自支撑电极是指形成自支撑膜的催化剂或生长在导电基质上的活性材料[133]。
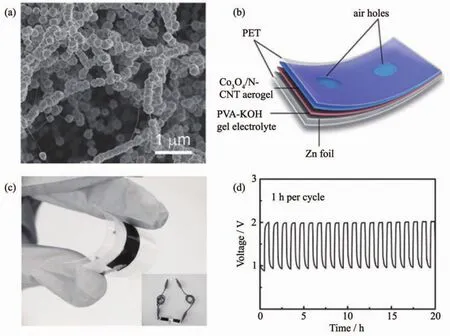
图10 (a)Co3O4/N-CNT的SEM图;(b)柔性锌-空电池示意图;(c)柔性、可逆全固态锌-空电池的光学照片,插图为2个并联的锌-空电池的点灯照片;(d)充放电循环性能[136]Fig.10 (a)SEM image of Co3O4/N-CNT;(b)Schematic diagram of flexible Zn-air battery;(c)Optical image of a flexible and rechargeable all-solid-state Zn-air battery,the inset shows two Zn-air batteries connected in series powering a red LED;(d)Charging and discharging cycling performance[136]
Cai等[134]合成了一种负载在石墨烯泡沫上的N掺杂碳纳米管阵列(N/VA-CNTs/GF)自支撑催化剂,石墨烯泡沫与碳纳米管阵列的结合同时赋予了材料优异的离子传输和气体扩散能力。Li等[135]以碳纳米管、石墨烯和Co(NO3)2为原料,通过自组装和氨气热处理制备了柔性自支撑的NG/CNT/Co3O4纸电极,表现出较高的ORR活性和稳定性。针对自支撑电极的测试除了常规电池所需的充放电和循环寿命等重要指标,扭曲和拉伸等作用下的电化学活性和稳定性至关重要。Li等[136]以柔性碳纳米管气凝胶为基础,经电化学活化在气凝胶缺陷部位引入亲水性官能团,再通过水热反应原位生长Co3O4颗粒,最终制备了Co3O4纳米粒子修饰的N-CNT气凝胶(Co3O4/N-CNT)(图10a)。该柔性、自支撑催化剂导电性能好,起始电势为0.90 V,接近Pt/C催化剂(0.94 V)。同时还表现出较好的析氧性能。如图10b所示,将锌板作为金属阳极,不加粘结剂与导电剂,以聚乙烯醇和KOH作为凝胶电解质,Co3O4/N-CNT作为空气电极组装成柔性锌-空电池(图10c),电荷转移阻力仅为4 Ω,循环充放电20 h后电池性能无明显衰减(图10d)。Chen等[137]结合电沉积和CVD法将一维NCNTs和介孔Co3O4纳米片阵列生长在不锈钢网上,得到头发状空气电极,该自支撑电极在不同扭曲状态下过电位无明显变化。显然,上述系列自支撑电极在可穿戴器件领域具有无可比拟的优势。不过,目前自支撑电极的活性和稳定性还需进一步提升。
表1为目前报道的典型碳纳米管基非贵金属催化剂的ORR性能对比。
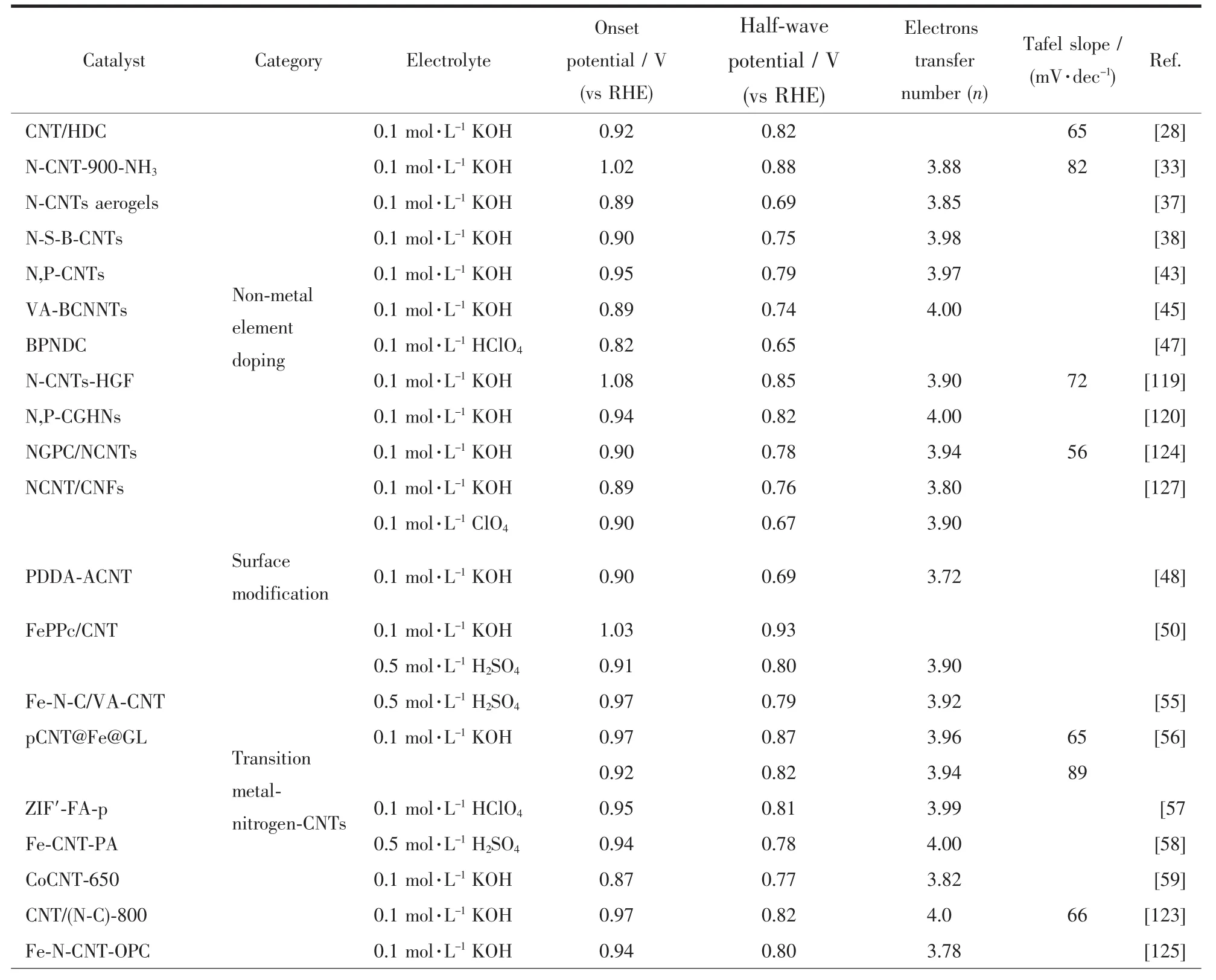
表1 典型碳纳米管基非贵金属催化剂的ORR性能Table 1 ORR performance of the typical CNTs-based noble-metal-free electrocatalysts
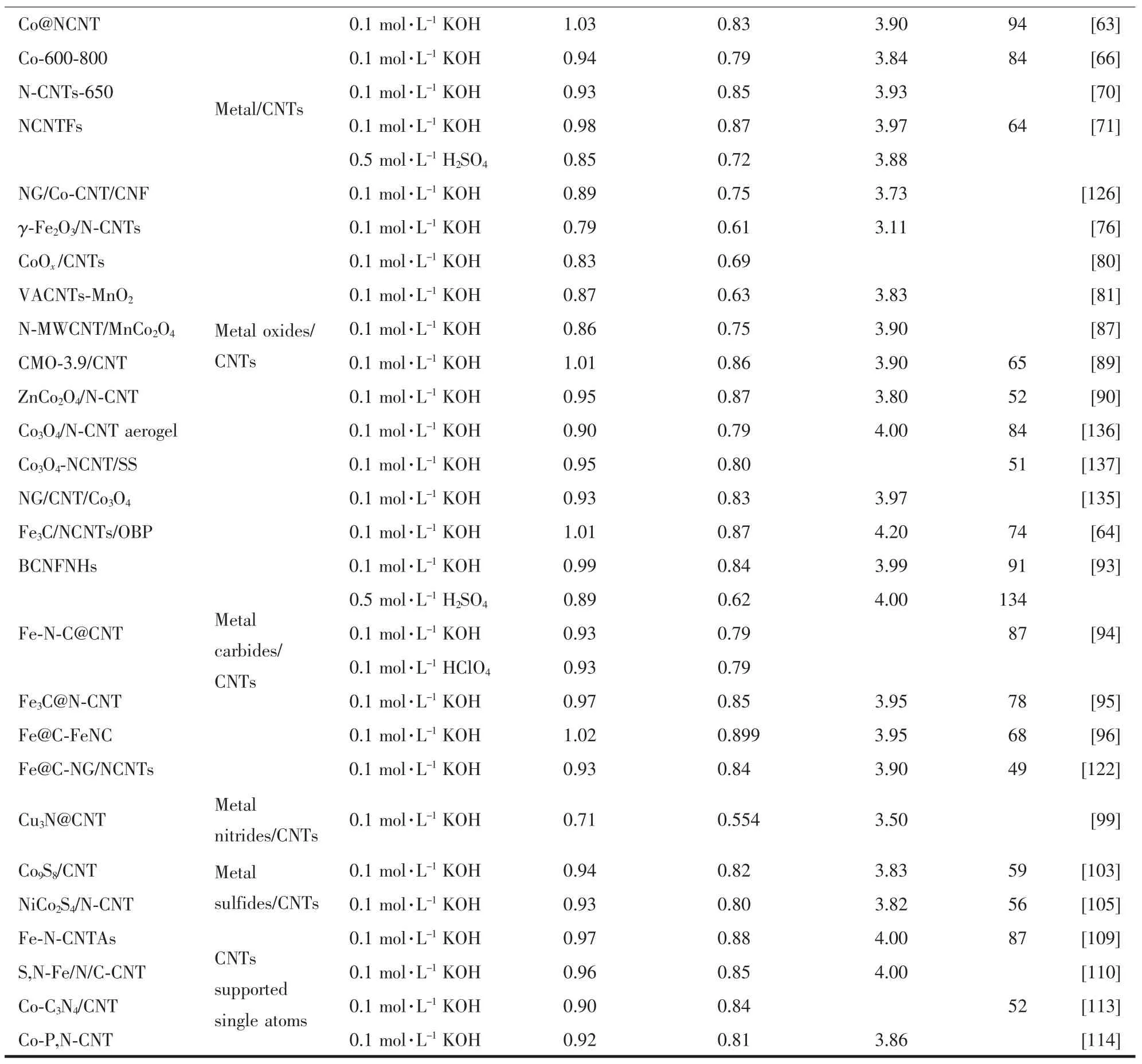
续表1
7 结论与展望
本文从非金属掺杂、过渡金属-氮-碳纳米管、碳纳米管负载过渡金属及其衍生物、碳纳米管负载单原子、与其他碳材料复合以及碳纳米管基自支撑电极等6方面综述了碳纳米管基ORR催化剂的研究进展。作为一类重要的非贵金属碳基电催化剂,碳纳米管ORR催化剂表现出了与商业Pt/C相当乃至更好的催化性能,在取代贵金属催化剂方面表现出巨大潜力,但仍有许多尚未解决的问题。笔者认为,今后的研究应着重以下方面:
(1)降低碳纳米管的制备成本和能耗。
(2)单原子催化剂中单原子负载量及催化剂的构-效关系。单原子具有高的比表面能,如何避免在制备过程中团聚的前提下进一步提高金属原子的负载量。单原子与传统负载型催化剂的活性材料配位环境不同,深入理解单原子催化体系的构-效关系是一项挑战。
(3)提高催化剂在酸性条件下的活性。探究催化活性与组成、电子性质、微观结构的构-效关系,结合理论计算在分子、原子水平确定活性位点,构筑高效、稳定的酸性碳纳米管基催化剂是ORR研究的重点方向之一。
(4)全电池性能测试。半电池测试评价催化性能时排除了扩散阻力对活性的干扰,将其组装成全电池测试应该是一个重要的补充手段。
(5)自支撑电极性能仍需提高。研究自支撑电极必须兼顾催化性能、柔性和可靠性。
致谢:感谢省部共建耐火材料与冶金国家重点实验室(武汉科技大学)开放基金(No.G201501)、高性能陶瓷和超微结构国家重点实验室开放课题(No.SKL201701SIC)和国防科技大学科研计划的资助。
参考文献:
[1]Stamenkovic V R,Strmcnik D,Lopes P P,et al.Nat.Mater.,2016,16:57-69
[2]Fu J,Cano Z P,Park M G,et al.Adv.Mater.,2017,29:1604685-1604718
[3]Liang Y Y,Li Y G,Wang H L,et al.Nat.Mater.,2011,10:780-786
[4]Zhou M,Wang H L,Guo S J.Chem.Soc.Rev.,2016,45:1273-1307
[5]Jaouen F,Proietti E,Lefèvre M,et al.Energy Environ.Sci.,2011,4:114-130
[6]Li Y G,Zhou W,Wang H L,et al.Nat.Nanotecnnol.,2012,7:394-400
[7]Wu N,Lei Y P,Wang Q C,et al.Nano Res.,2017,10:2332-2343
[8]Wang Q C,Chen Z Y,Wu N,et al.ChemElectroChem,2017,4:514-520
[9]Wu N,Wang Y D,Lei Y P,et al.Sci.Rep.,2015,5:17396-17404
[10]Lei Y P,Shi Q,Han C,et al.Nano Res.,2016,9:2498-2509
[11]Cai X Y,Lai L F,Lin J Y,et al.Mater.Horiz.,2017,4:945-976
[12]Ding Y L,Kopold P,Hahn K,et al.Adv.Funct.Mater.,2016,26:1112-1119
[13]Gu D,Li W,Wang F,et al.Angew.Chem.Int.Ed.,2015,54:7060-7064
[14]Chen Y G,Wang J J,Liu H,et al.J.Phys.Chem.C,2011,115:3769-3776
[15]Wu P,Cheng S,Yao M H,et al.Adv.Funct.Mater.,2017,27:1702160-1702168
[16]Zhao J,Su Y J,Yang Z,et al.Carbon,2013,58:92-98
[17]Das R,Shahnavaz Z,Ali M E,et al.Nanoscale Res.Lett.,2016,11:510-532
[18]Chung H T,Zelenay P.Chem.Commun.,2015,51:13546-13549
[19]Wen Z H,Ci S Q,Hou Y,et al.Angew.Chem.Int.Ed.,2014,53:6496-6500
[20]Chung H T,Won J H,Zelenay P.Nat.Commun.,2013,4:1922-1926
[21]Cheng Y,Zhang J,Jiang S P.Chem.Commun.,2015,51:13764-13767
[22]Yan Y B,Miao J W,Yang Z H,et al.Chem.Soc.Rev.,2015,44:3295-3346
[23]Wang X,Ouyang C B,Dou S,et al.RSC Adv.,2015,5:41901-41904
[24]Wu Z Y,Benchafia E M,Iqbal Z,et al.Angew.Chem.Int.Ed.,2014,126:12763-12767
[25]Gentil S,Serre D,Philouze C,et al.Angew.Chem.Int.Ed.,2015,55:2517-2521
[26]Wei P J,Yu G Q,Naruta Y,et al.Angew.Chem.Int.Ed.,2014,53:6659-6663
[27]Rao C V,Cabrera C R,Ishikawa Y.J.Phys.Chem.Lett.,2010,1:2622-2627
[28]Sa Y J,Park C,Jeong H Y,et al.Angew.Chem.Int.Ed.,2014,26:4186-4190
[29]Gao F,Zhao G L,Yang S Z.ACS Catal.,2014,4:1267-1273
[30]Gong K P,Du F,Xia Z H,et al.Science,2009,323:760-764
[31]Sharifi T,Hu G Z,Jia X E,et al.ACS Nano,2012,6:8904-8912
[32]Zhao A Q,Masa J,Schuhmann W,et al.J.Phys.Chem.C,2013,117:24283-24291
[33]Zhu X,Zhu Y H,Tian C C,et al.J.Mater.Chem.A,2017,5:4507-4512
[34]Wu G,Santandreu A,Kellogg W,et al.Nano Energy,2016,29:83-110
[35]Lee S,Choun M,Ye Y J,et al.Angew.Chem.Int.Ed.,2015,54:9230-9234
[36]Tian G L,Zhang Q,Zhang B S,et al.Adv.Funct.Mater.,2014,24:5956-5962
[37]Du R,Zhang N,Zhu J H,et al.Small,2015,11:3903-3908
[38]Liu S,Li G Z,Gao Y Y,et al.Catal.Sci.Technol.,2017,7:4007-4016
[39]Liu Z W,Shi Q Q,Zhang R F,et al.J.Power Sources,2014,268:171-175
[40]Yang L J,Jiang S J,Zhao Y,et al.Angew.Chem.Int.Ed.,2011,123:7270-7273
[41]Shi Q,Lei Y P,Wang Y D,et al.Curr.Appl.Phys.,2015,15:1606-1614
[42]Wang Y X,Lei Y P,Wang H P.RSC Adv.,2016,6:73560-73565
[43]Zhu J L,Jiang S P,Wang R H,et al.J.Mater.Chem.A,2014,2:15448-15453
[44]Zhao Y,Yang L J,Chen S,et al.J.Am.Chem.Soc.,2013,135:1201-1204
[45]Wang S Y,Iyyamperumal E,Roy A,et al.Angew.Chem.Int.Ed.,2011,50:11756-11760
[46]Shi Q Q,Peng F,Liao S X,et al.J.Mater.Chem.A,2013,1:14853-14857
[47]Choi C H,Park S H,Woo S I.ACS Nano,2012,6:7084-7091
[48]Wang S Y,Yu D S,Dai L M.J.Am.Chem.Soc.,2011,133:5182-5185
[49]Chen Y F,Liu Q,Wang J C.Nano Adv.,2016,1:79-89
[50]Wang X X,Wang B,Zhong J,et al.Nano Res.,2016,9:1497-1506
[51]Masa J,Xia W,Muhler M,et al.Angew.Chem.Int.Ed.,2015,54:10102-10120
[52]Zitolo A,Goellner V,Armel V,et al.Nat.Mater.,2015,14:937-944
[53]Jia Q Y,Ramaswamy N,Hafiz H,et al.ACS Nano,2015,9:12496-12505
[54]Sa Y J,Seo D J,Woo J,et al.J.Am.Chem.Soc.,2016,138:15046-15056
[55]Yasuda S,Furuya A,Uchibori Y,et al.Adv.Funct.Mater.,2016,26:738-744
[56]Ahn S H,Yu X W,Manthiram A.Adv.Mater.,2017,29:1606534-1606543
[57]Zhang C,Wang Y C,An B,et al.Adv.Mater.,2016,29:1604556-1604562
[58]Yang G,Choi W,Pu X,et al.Energy Environ.Sci.,2015,8:1799-1807
[59]Liu J H,Shen A L,Wei X F,et al.ACS Appl.Mater.Interface,2015,7:20507-20512
[60]Pan X L,Bao X H.Chem.Commun.,2008,47:6271-6281
[61]Liu Y Y,Jiang H L,Zhu Y H,et al.J.Mater.Chem.A,2016,4:1694-1701
[62]Xiao J W,Chen C,Xi J B,et al.Nanoscale,2015,7:7056-7064
[63]Zhu J B,Xiao M L,Liu C P,et al.J.Mater.Chem.A,2015,3:21451-21459
[64]Fu G T,Chen Y F,Cui Z M,et al.Nano Lett.,2016,16:6516-6522
[65]Deng D H,Yu L,Chen X Q,et al.Angew.Chem.Int.Ed.,2013,5:371-375
[66]Cao T,Wang D S,Zhang J T,et al.Chem.Eur.J.,2015,21:14022-14029
[67]Fu S F,Zhu C Z,Song J H,et al.Adv.Energy Mater.,2017,7:1700363-1700381
[68]Liu S H,Wang Z Y,Zhou S,et al.Adv.Mater.,2017,29:1700874-1700883
[69]You B,Jiang N,Sheng M L,et al.ACS Catal.,2015,5:7068-7076
[70]Meng J S,Niu C J,Xu L H,et al.J.Am.Chem.Soc.,2017,139:8212-8221
[71]Xia B Y,Yan Y,Li N,et al.Nat.Energy,2016,1:15006-15013
[72]Wang P W,Hayashi T,Meng Q A,et al.Small,2017,13:1601250-1601256
[73]Lu Z Y,Xu W W,Ma J,et al.Adv.Mater.,2016,28:7155-7161
[74]Chen Me J,Wang L,Yang H P,et al.J.Power Sources,2017,375:277-290
[75]Andersen N I,Serov A,Atanassov P.Appl.Catal.,B,2015,163:623-627
[76]Sun M,Zhang G,Liu H J,et al.Sci.China Mater.,2015,58:683-692
[77]Yang Z,Zhou X,Nie H,et al.ACS Appl.Mater.Interface,2011,3:2601-2606
[78]Sun M,Dong Y Z,Zhang G,et al.J.Mater.Chem.A,2014,2:13635-13640
[79]Li Y G,Gong M,Liang Y Y,et al.Nat.Commun.,2013,4:1805-1811
[80]Seo B,Sa Y J,Woo J,et al.ACS Catal.,2016,6:4347-4355
[81]Yang Z,Zhou X M,Jin Z P,et al.Adv.Mater.,2014,26:3156-3161
[82]Zhao Q,Yan Z H,Chen C C,et al.Chem.Rev.,2017,117:10121-10211
[83]Zhao A Q,Masa J,Xia W,et al.J.Am.Chem.Soc.,2014,136:7551-7554
[84]Zhang H,Qiao H,Wang H Y,et al.Nanoscale,2014,6:10235-10242
[85]Li P X,Ma R G,Zhou Y,et al.RSC Adv.,2015,5:73834-73841
[86]Yan W N,Bian W Y,Jin C,et al.Electrochim.Acta,2015,177:65-72
[87]Yuvaraj S,Vignesh A,Shanmugam S,et al.Int.J.Hydrogen Energy,2016,41:15199-15207
[88]Li J X,Zou M Z,Wen W W,et al.J.Mater.Chem.A,2014,2:10257-10262
[89]Shi J J,Lei K X,Sun W Y,et al.Nano Res.,2017,10:3836-3847
[90]Liu Z Q,Cheng H,Li N,et al.Adv.Mater.,2016,28:3777-3784
[91]Li J S,Li S L,Tang Y J,et al.Chem.Commun.,2015,51:2710-2713
[92]Xiao Y,Hwang J Y,Sun Y K.J.Mater.Chem.A,2016,4:10379-10392
[93]Yang W X,Liu X J,Yue X Y,et al.J.Am.Chem.Soc.,2015,137:1436-1439
[94]Zhang Y,Jiang W J,Guo L,et al.ACS Appl.Mater.Interface,2015,7:11508-11515
[95]Guan B Y,Yu L,Lou X W.Energy Environ.Sci.,2016,9:3092-3096
[96]Jiang W J,Gu L,Li L,et al.J.Am.Chem.Soc.,2016,138:3570-3578
[97]Pang M,Li C,Ding L,et al.Ind.Eng.Chem.Res.,2010,49:4169-4174
[98]Xie J F,Xie Y.Chem.Eur.J.,2016,22;3588-3598
[99]Su C Y,Liu B H,Lin T J,et al.J.Mater.Chem.A,2015,3:18983-18990
[100]Youn D H,Bae G H,Han S,et al.J.Mater.Chem.A,2013,1:8007-8015
[101]Dong S M,Chen X,Zhang K J,et al.Chem.Commun.,2011,47:11291-11293
[102]Yu X Y,Lou X W.Adv.Energy Mater.,2018,8:1701592
[103]Li H,Guo Z,Wang X W.J.Mater.Chem.A,2017,5:21353-21361
[104]Periasamy A P,Wu W P,Lin G L,et al.J.Mater.Chem.A,2014,2:11899-11904
[105]Han X P,Wu X Y,Zhong C,et al.Nano Energy,2017,37:541-550
[106]Bayatsarmadi B,Zheng Y,Vasileff A,et al.Small,2017,13:1700191-1700209
[107]Chen Y J,Ji S F,Wang Y G,et al.Angew.Chem.Int.Ed.,2017,129:7041-7046
[108]Zhu C Z,Fu S F,Shi Q R,et al.Angew.Chem.Int.Ed.,2017,56:13944-13960
[109]Zhu C Z,Fu S F,Song J H,et al.Small,2017,13:1603407-1603413
[110]Chen P Z,Zhou T P,Xing L L,et al.Angew.Chem.Int.Ed.,2016,56:610-615
[111]Zhang J S,Chen Y,Wang X C.Energy Environ.Sci.,2015,8:3092-3108
[112]Han C,Wang Y D,Lei Y P,et al.Nano Res.,2015,8:1199-1209
[113]Zheng Y,Jiao Y,Zhu Y H,et al.J.Am.Chem.Soc.,2017,139:3336-3339
[114]Guo S Y,Yuan P F,Zhang J N,et al.Chem.Commun.,2017,53:9862-9865
[115]Chen P,Xiao T Y,Qian Y H,et al.Adv.Mater.,2013,25:3192-3196
[116]Peng S J,Li L L,Han X P,et al.Angew.Chem.Int.Ed.,2014,5:12594-12599
[117]Chen S,Duan J J,Jaroniec M,et al.Adv.Mater.,2014,26:2925-2930
[118]Wang H,Jia J,Song P F,et al.Angew.Chem.Int.Ed.,2017,56:7847-7852
[119]Cheng Z H,Fu Q,Li C X,et al.J.Mater.Chem.A,2016,4:18240-18248
[120]Yang J,Sun H Y,Liang H Y,et al.Adv.Mater.,2016,28:4606-4613
[121]Zhou X M,Tian Z M,Li J,et al.Nanoscale,2014,6:2603-2607
[122]Wang Q C,Lei Y P,Chen Z Y,et al.J.Mater.Chem.A,2018,6:516-526
[123]Li J C,Zhao S Y,Hou P X,et al.Nanoscale,2015,7:19201-19206
[124]Zhang L J,Wang X Y,Wang R H,et al.Chem.Mater.,2015,27:7610-7618
[125]Liang J,Zhou R F,Chen X M,et al.Adv.Mater.,2014,26:6074-6079
[126]Shi Q,Wang Y D,Wang Z M,et al.Nano Res.,2016,9:317-328
[127]Guo Q H,Zhao D,Liu S W,et al.Electrochim.Acta,2014,138:318-324
[128]Tan P,Chen B,Xu H R,et al.Energy Environ.Sci.,2017,10:2056-2080
[129]Liu Q,Wang Y B,Dai L M,et al.Adv.Mater.,2016,28:3000-3006
[130]Su C Y,Cheng H,Li W,et al.Adv.Energy Mater.,2017,7:1602420-1602431
[131]Cheng H,Chen J M,Li Q J,et al.Chem.Commun.,2017,53:11596-11599
[132]Yu M H,Wang Z K,Hou C,et al.Adv.Mater.,2017,29:1602868-1602874
[133]Ma T Y,Dai S,Qiao S Z.Mater.Today,2016,19:265-273
[134]Cai X Y,Xia B Y,Franklin J,et al.J.Mater.Chem.A,2017,5:2488-2495
[135]Li S S,Cong H P,Wang P,et al.Nanoscale,2014,6:7534-7541
[136]Zeng S,Chen H Y,Wang H,et al.Small,2017,13:1700518-1700525
[137]Fu J,Hassan F M,Li J D,et al.Adv.Mater.,2016,28:6421-6428
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
防爆电机(2020年4期)2020-12-14
河北理科教学研究(2020年1期)2020-07-24
浙江大学学报(工学版)(2016年9期)2016-06-05
原子与分子物理学报(2015年3期)2015-11-24
高中生学习·高二版(2014年5期)2014-07-03
郑州大学学报(理学版)(2014年4期)2014-03-01
中国医学科学院学报(2010年4期)2010-03-25
