
细胞外超氧化物歧化酶DNA甲基化对动脉粥样硬化的诱导作用及作用靶点
2018-04-10 02:47陈佳杨晓龙张旭峰
中国现代医学杂志 2018年10期
陈佳,杨晓龙,张旭峰
(上海中医药大学附属曙光医院1.急诊科,2.重症医学科,3.检验科,上海 201203)
动脉粥样硬化(Atherosclerosis,AS)是导致心肌梗死、心绞痛、冠状动脉粥样硬化性心脏病(冠心病)、脑梗死等心脑血管疾病的主要原因[1-3]。目前研究已证实氧化应激诱导血管炎症改变,引起的局部血管炎症反应和细胞过度增殖参与了AS的发生和进展。超氧化物歧化酶(superoxide dismutase,SOD)是机体抵抗氧自由基损伤的主要防御体系,其中细胞外SOD(extracellular superoxide dismutase,EC-SOD)是唯一在细胞外分布并发挥抗氧化功能的酶[4-6],也是血管壁抗氧化酶的主要来源。DNA甲基化作为一种重要的表观遗传修饰方式,参与了AS的发病和局部炎症反应。尽管如此,目前依然无法确定长期持续性血脂异常所诱导的表观遗传改变,特别是EC-SOD甲基化,是否与AS的发病有关。鉴于此,本研究通过复制高脂饮食纯合子载脂蛋白E基因敲除(apolipoprotein E-knockout,ApoE-/-)小鼠模型和THP-1单核细胞源性巨噬细胞,观察EC-SOD甲基化与AS的关系及可能作用靶点,旨在为AS的防治提供理论基础。
1 材料与方法
1.1 试剂与抗体
RPMI-1640培养基购自美国Gibco公司,胰蛋白酶、二甲基亚砜、胎牛血清、BCA蛋白浓度测定试剂盒购自广州碧云天生物技术研究所,Trizol试剂、逆转录聚合酶链反应(reverse transcriptionpolymerase chain reaction,RT-PCR)试剂盒、脂质体LipofectamineTM2000购自美国Invitrogen公司,pEGFP-N1-DNMT1重组质粒委托上海捷瑞生物工程有限公司合成,DNA提取试剂盒、MMLV逆转录酶、dNTP、Taq DNA聚合酶、琼脂糖购自美国Promega公司,THP-1单核细胞株、大肠杆菌DH5α购自上海抚生实业有限公司,基因引物由上海生工公司合成,丙烯酰胺、PVDF膜、十二烷基硫酸钠购自美国Sigma公司。DNA甲基转移酶1(DNA methyltransferase 1,DNMT1)、SOD3抗体、β-actin小鼠单克隆抗体、异硫氰酸荧光素(fluorescein isothiocyanate,FITC)标记的羊抗兔子IgG二抗购自美国Santa Cruz公司,丙二醛(Malonaldehyde,MDA)、活性氧(reactive oxygen species,ROS)检测试剂盒均购自上海心语生物科技有限公司。
1.2 动物分组与处理
16只雄性ApoE-/-实验小鼠(ApoE-/-组)和16只C57BL/6小鼠(WT组)购自北京大学实验动物中心[许可证号:SYXK(京)2016-0028],8周龄,体重18~22 g,标准化饲养房饲养[SPF级动物房,12 h光照与12 h黑暗循环,相对湿度40%~70%,温度(22±2)℃]。普通饲料配方:麸皮25%,玉米25%,豆饼13%,大麦20%,鱼粉6%,其他辅料11%;高脂饲料配方:在普通饲料中加入5%脂肪、1.5%胆固醇、5%蛋黄粉和0.2%胆酸钠。WT组给予不含任何高脂成分的普通饲料喂养,ApoE-/-组给予高脂饲料喂养,小鼠自由摄食和饮水20周后,进行后续实验。
1.3 细胞培养、分组与处理
THP-1单核细胞株接种于RPMI-1640培养基,于37℃、5%CO2恒温培养箱中培养,光学显微镜下观察细胞形态,当细胞贴壁生长、且出现伪足时,提示已经分化为巨噬细胞。将细胞分为3组:空白对照组(NC组)、空白质粒组(pEGFP-N1组)和重组质粒组(pEGFP-N1-DNMT1组);取分化为巨噬细胞的THP-1细胞株,NC组用含15%胎牛血清的RPMI-1640培养基继续培养,pEGFP-N1组加入10μl脂质体Lipofectamine和20μg空质粒载体pEGFP-N1,pEGFP-N1-DNMT1组加入10μl脂质体Lipofectamine和20μg pEGFP-N1-DNMT1重组质粒载体,于37℃、5%CO2下培养,每6 h更换1次培养液,各组细胞继续培养24 h后,进行后续实验。
1.4 动物处理与取样
两组于喂养第8、12、16、20周时,每次每组取4只小鼠,腹腔注射2 ml/100 g麻醉,置于动物手术台上,摘除眼球,取眼眶静脉血3 ml,静置20 min,5 000 r/min离心15 min,吸取上清液,置于-20℃冰箱保存待检。再将小鼠处死,开腹取出主动脉,生理盐水冲洗干净,切取距主动脉弓0.5 cm处的主动脉,分为2部分,一部分采用4%多聚甲醛固定,其余部分于液氮中保存。
1.5 检测指标
1.5.1血脂检测 采用全自动生化分析仪检测小鼠低密度脂蛋白(low density lipoprotein,LDL)、高密度脂蛋白(high density lipoprotein,HDL)、三酰甘油(Triglyceride,TG)、总胆固醇(total cholesterol,TC)等血脂水平,其中LDL、HDL采用常规酶法,TG、TC采用终点法。
1.5.2组织形态学观察取4%多聚甲醛固定的主动脉组织,石蜡包埋,制成4μm的连续切片,采用苏木素-伊红染色,中性树胶封片,光学显微镜下观察组织形态学特征。
1.5.3氧化应激指标检测取4μg组织样本,匀浆器中打成匀浆,12 000 r/min离心15 min,取上清液待测。采用分光光度比色法检测主动脉组织MDA、ROS含量,所有样本均于酶标仪上检测560 nm处吸光度值,采用标准曲线计算出待测样品蛋白的浓度。
1.5.4RT-PCR检测主动脉组织EC-SOD、DNMT1 mRNA水平液氮中取出主动脉组织,自然解冻,Trizol RNA提取试剂盒提取组织总RNA,紫外分光光度法检测总RNA浓度。将1μg总RNA按照GeneAmp PCR试剂盒说明书逆转录成cDNA;合成体系:逆转录缓冲液3μl,RNA逆转录模板1μl,逆转录酶2μl,正向、反向引物各1μl,加入双蒸水补充至总反应体系为20μl,37℃金属浴反应50 min合成cDNA。将cDNA置于PCR反应体系中进行扩增(引物序列见附表)。PCR反应体系:PCR试剂混合液12.5μl,DEPC 8μl,cDNA 2.5μl,正反向引物 1μl,总反应体系25μl;反应条件:95℃预变性1 min,95℃变性30 s,55℃退火30 s,72℃延伸1 min,40个循环后72℃ 5 min,溶解曲线条件为55℃~95℃,将PCR产物置于琼脂糖凝胶电泳分离扩增片段,检测基因荧光信号强度值(Ct),计算基因表达量(2-△△Ct),以β-actin作为内参照进行校正,分析目的基因相对表达量。

附表 引物序列及扩增片段长度
1.5.5巢式甲基化特异性PCR检测主动脉组织ECSOD DNA甲基化程度DNA提取试剂盒提取组织总RNA,4%琼脂糖凝胶检测基因组DNA浓度和纯度。PCR反应体系:Power SYBR Green PCR Master Mix10μl,引物 1μl,DNA 模板 2.5μl,ddH2O 15μl。第1轮PCR采用外侧引物,反应条件:95℃预变性5 min,97℃变性30 s,56℃退火30 s,总计35个循环。将第1轮PCR产物稀释50倍,取1μl进行第2轮PCR反应,95℃预变性5 min,97℃变性30 s,62℃退火15 s,总计35个循环。阴性对照为人外周血淋巴细胞,将PCR产物采用琼脂糖凝胶电泳,溴化乙锭染色,紫外灯下观察结果,甲基化%=甲基化光密度值/(甲基化光密度值+非甲基化光密度值)×100%。
1.5.6Western blot检测EC-SOD、DNMT1蛋白水平和EC-SOD DNA甲基化程度取出对数生长期细胞,弃去培养液,PBS冲洗,加入离心管中,再加入1 ml细胞裂解液置于冰浴上裂解30 min,4℃条件下离心15 min,采用Western blot检测EC-SOD、DNMT1蛋白水平,将20μg蛋白提取液置于10%SDS-PAGE电泳分离,常规湿法转膜,加入5%脱脂牛奶孵育封闭2 h。加入鼠抗人EC-SOD单克隆抗体、兔抗人DNMT1单克隆抗体,4℃孵育24 h。再滴加FITC标记的二抗37℃孵育2 h。PBS冲洗3次,按照ECL化学发光显影试剂盒显影,以β-actin作为内参照,分析目的条带相对表达量。各组细胞EC-SOD DNA甲基化程度方法同上。
1.6 统计学方法
采用SPSS19.0软件进行数据处理,计量资料用均数±标准差(±s)表示,采用单因素方差分析,两两比较采用SNK-q检验,P<0.05为差异有统计学意义。
2 结果
2.1 血脂水平变化
第 8、12、16、20周,ApoE-/-组 TC(见图 1A)、LDL(见图1B)、TG(见图1C)呈升高趋势(F=3.140、4.183 和 4.920,P=0.041、0.033 和 0.029),HDL(见图1D)无明显变化(F=0.735,P=0.955);各时段ApoE-/-组TC、LDL、TG高于WT组,HDL低于WT组(P<0.05)。
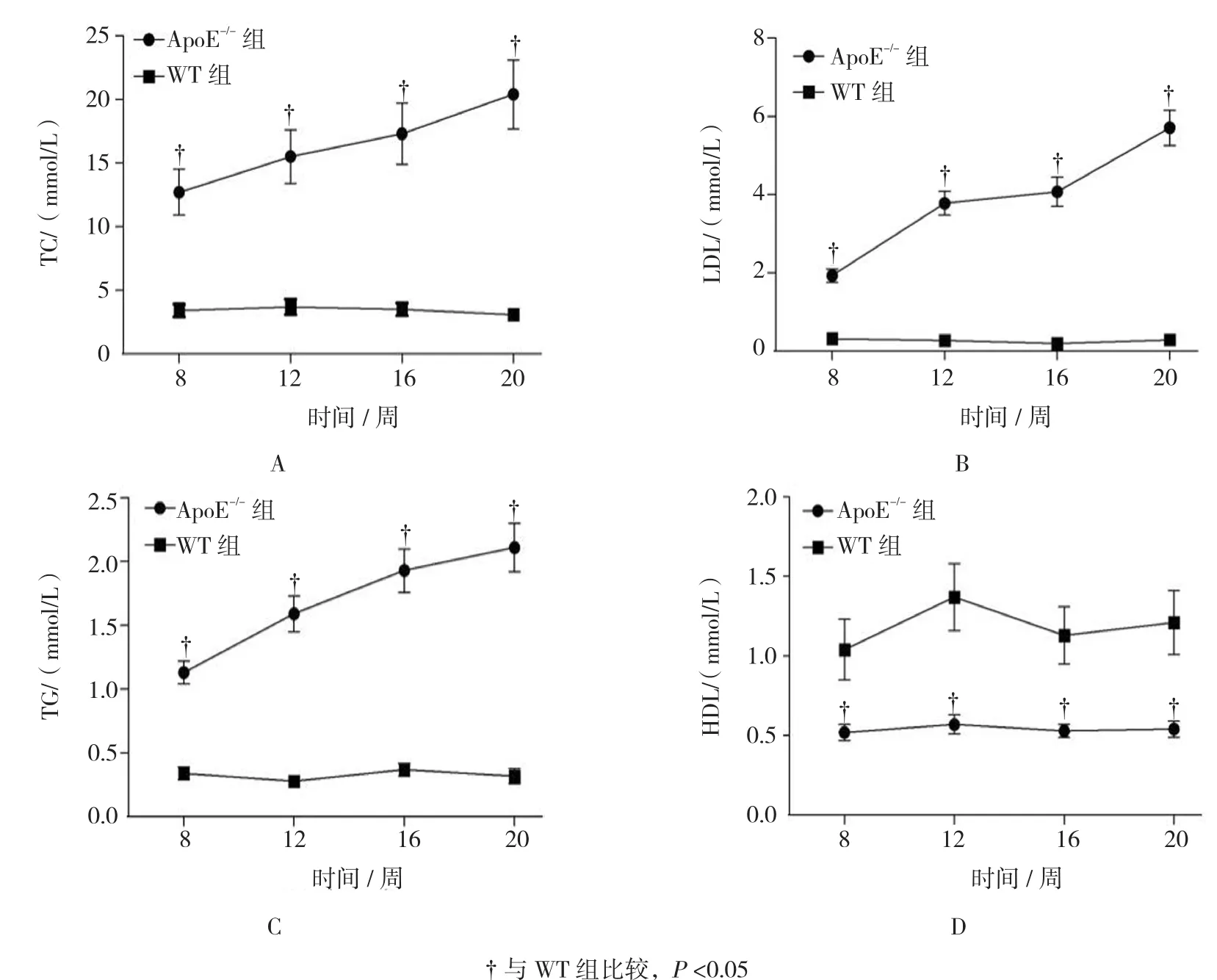
图1 两组血脂比较
2.2 主动脉病理形态变化
第16、20周,WT组动脉组织内皮细胞结构规则有序,平滑肌细胞排列整齐,内皮下无炎症细胞或脂质沉积;ApoE-/-组主动脉内膜明显增厚,血管壁伴有弥漫性隆起,平滑肌细胞排列无序,内皮下伴有炎症浸润和脂质沉积。见图2。
2.3 氧化应激指标变化
第 8、12、16、20周,ApoE-/-组MDA(见图 3A)、ROS(见图3B)呈升高趋势(F=8.143和9.224,P=0.021和0.019),各时段ApoE-/-组MDA、ROS高于 WT 组(P<0.05)。
2.4 EC-SOD、DNMT1 mRNA及 EC-SOD甲基化水平变化
第 8、12、16、20 周,ApoE-/-组 EC-SOD mRNA(见图4A)表达水平低于WT组(F=17.320,P=0.000),DNMT1 mRNA表达水平(见图4B)和EC-SOD甲基化水平(见图4C)高于WT组(F=16.477和4.965,P=0.001和 0.024)。
2.5 pEGFP-N1-DNMT1转 染 后EC-SOD、DNMT1蛋白及EC-SOD甲基化水平变化
pEGFP-N1-DNMT1组DNMT1蛋白表达(见图5A)及EC-SOD甲基化水平(见图5C)高于pEGFPN1组(F=7.884和6.329,P=0.028和0.037)和NC组(F=7.955和6.059,P=0.004和0.019),EC-SOD蛋白表达水平(见图5B)低于pEGFP-N1组(F=15.365,P=0.000)和NC 组(F=6.331,P=0.037),pEGFP-N1组和NC组EC-SOD、DNMT1蛋白表达水平及ECSOD甲基化水平比较差异无统计学意义(P>0.05)。
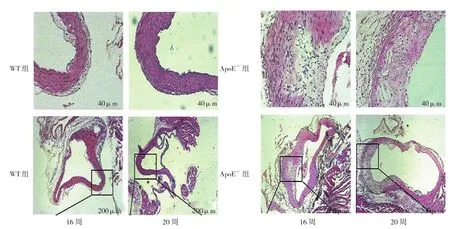
图2 两组主动脉病理形态变化
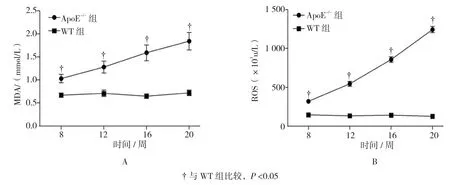
图3 两组氧化应激指标比较

图5 pEGFP-N1-DNMT1转染后EC-SOD、DNMT1蛋白表达及EC-SOD甲基化水平比较
3 讨论
正常情况下,机体处于氧化和抗氧化动态平衡,当平衡被打破后就会导致氧自由基生成、清除失衡,引起ROS蓄积,最终引起氧化应激损伤[7-9]。目前已证实氧化应激损伤与AS的发生和进展密切相关,氧化应激能通过诱导氧化作用,引起局部炎症反应和细胞增殖,因此针对性的抗氧化治疗成为AS防治的主要手段。ApoE-/-实验小鼠是复制AS模型常用的动物,本研究利用高脂饲料喂养ApoE-/-小鼠,结果显示ApoE-/-组TC、LDL、TG高于WT组,HDL低于WT组;主动脉根部切片染色显示ApoE-/-组主动脉内膜明显增厚,WT组动脉组织内皮细胞结构规则有序,证实AS小鼠模型复制成功。进一步分析两组小鼠的氧化应激指标,结果显示ApoE-/-组MDA、ROS呈升高趋势,各时段ApoE-/-组MDA、ROS高于WT组,说明AS与氧化应激损伤密切相关。NOMIYA等[10]报道称氧化应激能损伤血管内皮细胞,进而诱发粥样斑块形成和脂质沉积,这一过程亦是AS的始动因素。PEREIRA等[11]也证实高脂喂养的ApoE-/-小鼠血脂异常升高,ROS水平增加,证实高脂血症也会诱导氧化应激反应。
机体发生氧化应激时,能够启动自身的抗氧化防御体系,其中EC-SOD唯一分布于细胞外的抗氧化酶,其通过与硫酸乙酰肝素结合,将氧自由基歧化为H2O2,保护组织避免发生自由基损伤,预防AS发生[12-13]。最新研究发现[14],EC-SOD还具有调节内源性NO的作用,所产生的NO能够扩张血管平滑肌、抑制血小板聚集,进而抑制AS发生和进展。本研究发现ApoE-/-组EC-SOD mRNA表达水平低于WT组,说明EC-SOD含量偏低引起的抗氧化防御体系功能减弱是导致AS的诱因之一。TEOH-FITZGERALD等[15]报道称细胞外高浓度EC-SOD能够有效清除氧自由基,减少超氧化亚硝基阴离子形成,进而保护细胞避免氧自由基损害,抑制AS的进程。BREAK等[16]也证实EC-SOD能够抑制还原型辅酶Ⅱ(NADPH)和ROS增多而产生的氧自由基,将小鼠EC-SOD基因敲除后,小鼠发生AS的风险升高。
目前已经明确EC-SOD是抗氧化的主要酶系,但是其在粥样硬化过程中的作用机制仍存在一定争议。随着分子生物学的研究深入,人们逐渐认识到表观遗传在心血管疾病发病中的作用。作为表观遗传的典型标记,DNA甲基化在多种生物学功能中发挥着重要作用[17-18]。杨程等[19]对40例确诊为AS的患者外周血基质金属蛋白酶组织抑制剂1(TIMP-1)进行检测,结果发现AS患者外周血TIMP-1含量低于正常对照组,进一步发现其降低的机制与TIMP-1基因启动子区的高甲基化密切相关,提示基因DNA甲基化也是导致AS形成的原因之一。本研究ApoE-/-组EC-SOD甲基化水平高于WT组,说明AS发病可能与EC-SOD甲基化水平增加,进而导致EC-SOD表达下调有关。DNMT1是维持基因DNA甲基化修饰状态的主要酶系,也是调节基因DNA甲基化水平的关键[20];DNMT1表达与DNA甲基化程度正相关,与基因表达负相关[21]。本研究证实ApoE-/-组DNMT1表达水平高于WT组,成功转染DNMT1的巨噬细胞DNMT1蛋白表达及EC-SOD甲基化升高,EC-SOD蛋白表达降低,说明DNMT1催化EC-SOD转甲基反应,导致EC-SOD水平降低,进而加重氧化应激损伤,最终形成AS。这一结果也提示DNMT1可以作为AS靶向治疗的1个潜在作用靶点。
综上所述,EC-SOD甲基化程度升高,导致ECSOD表达降低,机体抵抗氧化应激反应能力减弱,最终发展成AS。
参 考 文 献:
[1]PFENNIGER A, CHANSON M, KWAK B R. Connexins in atherosclerosis[J]. Biochimica Et Biophysica Acta, 2013, 1828(1):157-166.
[2]JR Y A, OHMI T, MONMA R, et al. Correlation between the characteristics of acceleration and visco elasticity of artery wall under pulsatile flow conditions (physical meaning of I*as a parameter of progressive behaviors of atherosclerosis and arteriosclerosis)[J]. Bio-Medical Materials and Engineering, 2013,23(1-2): 75-91.
[3]PEIFFER V, BHARATH A A, SHERWIN S J, et al. A novel method for quantifying spatial correlations between patterns of atherosclerosis and hemodynamic factors[J]. Journal of Biomechanical Engineering, 2013, 135(2): 124-130.
[4]SCAVENIUS C, PETERSEN J S, THOMSEN L R, et al. Murine extracellular superoxide dismutase is converted into the inactive fold by the Ser195Cys mutation[J]. Biochemistry, 2013, 52(19):3369-3375.
[5]SONNI S, LIOUTAS V A, SELIM M H. Epigenetic regulation of extracellular-superoxide dismutase in human monocytes[J]. Free Radical Biology & Medicine, 2013, 61(1): 197-205.
[6]BRESSLER J, SHIMMIN L C, BOERWINKLE E, et al. Global DNA methylation and risk of subclinical atherosclerosis in young adults: the pathobiological determinants of atherosclerosis in youth(PDAY) study[J]. Atherosclerosis, 2011, 219(2): 958-962.
[7]TSUCHIYA K, WESTERTERP M, MURPHY A J, et al. Expanded granulocyte/monocyte compartment in myeloid-speci fi c triple foxo knockout increases oxidative stress and accelerates atherosclerosis in mice[J]. Circulation Research, 2013, 112(7): 992-1003.
[8]DELGADO-ROCHE L, ACOSTA E, SOTO Y, et al. The treatment with an anti-glycosaminoglycan antibody reduces aortic oxidative stress in a rabbit model of atherosclerosis[J]. Free Radical Research, 2013, 47(4): 309-315.
[9]NYYSSÖNEN K, KURL S, KARPPI J, et al. LDL oxidative modification and carotid atherosclerosis: results of a multicenter study[J]. Atherosclerosis, 2012, 225(1): 231-236.
[10]NOMIYA M, SAGAWA K, YAZAKI J, et al. Increased bladder activity is associated with elevated oxidative stress markers and proinflammatory cytokines in a rat model of atherosclerosisinduced chronic bladder ischemia[J]. Neurourology and Urodynamics, 2012, 31(1): 185-189.
[11]PEREIRA S S, TEIXEIRA L G, AGUILAR E C, et al. Differences in adipose tissue in fl ammation and oxidative status in C57BL/6 and ApoE-/- mice fed high fat diet[J]. Animal Science Journal,2012, 83(7): 549-555.
[12]DIVYYA S, NAUSHAD S M, MURTHY P V, et al. GCPII,modulates oxidative stress and prostate cancer susceptibility through changes in methylation of RASSF1, BNIP3, GSTP1, and Ec - SOD[J]. Molecular Biology Reports, 2013, 40(10): 5541-5550.
[13]MAKINO J, KAMIYA T, HARA H, et al. TPA induces the expression of EC-SOD in human monocytic THP-1 cells:involvement of PKC, MEK/ERK and NOX-derived ROS[J]. Free Radical Research, 2012, 46(5): 637-644.
[14]AHMED M N, CODIPILLY C, HOGG N, et al. The protective effect of overexpression of extracellular superoxide dismutase on nitric oxide bioavailability in the lung after exposure to hyperoxia stress[J]. Experimental Lung Research, 2011, 37(1): 10-17.
[15]TEOH-FITZGERALD M L, FITZGERALD M P, JENSEN T J, et al. Genetic and epigenetic inactivation of extracellular superoxide dismutase promotes an invasive phenotype in human lung cancer by disrupting ECM homeostasis[J]. Molecular Cancer Research,2012, 10(1): 40-51.
[16]BREAK T J, JUN S, INDRAMOHAN M, et al. Extracellular superoxide dismutase inhibits innate immune responses and clearance of an intracellular bacterial infection[J]. Journal of Immunology, 2012, 188(7): 3342-3350.
[17]CHEN C, LI B. Chemiluminescence resonance energy transfer biosensing platform for site-specific determination of DNA methylation and assay of DNA methyltransferase activity using exonuclease III-assisted target recycling amplification[J].Biosensors & Bioelectronics, 2014, 54(8): 48-54.
[18]WEINER S A, GALBRAITH D A, ADAMS D C, et al. A survey of DNA methylation across social insect species, life stages, and castes reveals abundant and caste-associated methylation in a primitively social wasp[J]. The Science of Nature, 2013, 100(8):795-799.
[19]杨程, 田珏, 蔡欣, 等. 动脉粥样硬化患者基质金属蛋白酶组织抑制剂1基因DNA高甲基化的研究[J]. 实用医学杂志,2013, 29(21): 3530-3532.
[20]ZAMPIERI M, GUASTAFIERRO T, CALABRESE R, et al.ADP-ribose polymers localized on Ctcf-Parp1-DNMT1 complex prevent methylation of Ctcf target sites[J]. Biochemical Journal,2012, 441(2): 645-652.
[21]TAKESHITA K, SUETAKE I, YAMASHITA E, et al. Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (DNMT1)[J]. Proceedings of the National Academy of Sciences, 2011, 108(22): 9055-9059.
猜你喜欢
上海交通大学学报(医学版)(2022年3期)2022-05-05
昆明医科大学学报(2022年2期)2022-03-29
心肺血管病杂志(2020年5期)2021-01-14
世界科学技术-中医药现代化(2020年2期)2020-07-25
中国医学影像技术(2019年7期)2019-08-01
中成药(2018年6期)2018-07-11
医学研究杂志(2015年12期)2015-06-10
中国医科大学学报(2015年10期)2015-03-01
癌变·畸变·突变(2015年3期)2015-02-27
癌变·畸变·突变(2015年3期)2015-02-27
