
α-硫辛酸对糖尿病大鼠肾脏的保护作用及其可能的机制*
2018-04-10 02:47张小欢毛彦稳彭伟王圆圆刘丽荣刘玲伶石明隽肖瑛汤磊郭兵
中国现代医学杂志 2018年10期
张小欢 ,毛彦稳 ,彭伟 ,王圆圆 ,刘丽荣 ,刘玲伶 ,石明隽,肖瑛,汤磊,郭兵
(1.贵州医科大学 病理生理学教研室,贵州 贵阳 550025;2.贵州医科大学 重大疾病发病机制及药物防治特色重点实验室,贵州 贵阳 550025;3.贵州医科大学附属医院检验科,贵州 贵阳 550004;4.贵州医科大学 药学院,贵州 贵阳 550004)
糖尿病肾病(diabetic nephropathy,DN)是糖尿病(diabetes mellitus,DM)最常见的严重微血管并发症之一,DN晚期的主要病理特征包括肾小球硬化和肾小管间质纤维化。而氧化应激(oxidative stress,OS)在DN的发病过程中发挥着重要作用[1]。在正常生理状态下,适量的活性氧(reactive oxygen species,ROS)能迅速被肾组织内抗氧化物质(如超氧化物歧化酶、过氧化氢酶等)清除;在糖尿病或高血糖环境时,ROS产生增多而清除减少,体内聚集大量的ROS便可诱导肾脏固有细胞发生氧化应激,产生大量过氧化代谢产物(如丙二醛等);并且可以诱导胞内相关信号通路的激活,经核内转录因子介导促纤维化生长因子(如转化生长因子、结缔组织生长因子等)基因转录和高表达,最终出现细胞外基质(extracellular matrix,ECM)过度沉积而形成肾脏纤维化[2-3]。而转化生长因子(transforming growth factor β1,TGF-β1)信号通路是目前公认的与糖尿病肾脏纤维化密切相关的信号通路,主要通过诱导肾小管上皮细胞向间充质细胞转化(epithelial mesenchy-mal transition,EMT),促使ECM合成增加并抑制其降解而过度沉积,从而造成广泛的肾脏纤维化[4]。而核转录共抑制因子(Skirelated novel protein,SnoN)是 TGF-β1信号通路的负性调控因子,可以通过抑制TGF-β1信号通路来延缓肾脏纤维化进程。研究表明[5],α-硫辛酸(alpha lipoic acid,ALA)是一种含硫的抗氧化剂,可以清除活性氧和自由基,对氧化应激引起的组织损伤有治疗作用。因此,本研究旨在观察抗氧化剂ALA对DM大鼠肾脏的保护作用,并探讨其是否通过调节SnoN表达和TGF-β1信号通路来发挥对肾脏的保护作用,以进一步了解ALA的作用机制,为DN的防治提理论依据。
1 材料与方法
1.1 材料
1.1.1实验动物健康清洁级雄性Sprague-Dawley大鼠,体重(180±20)g,共24只;由北京华阜康生物科技股份有限公司提供,批号为SCXK(京)2009-0004。
1.1.2主要试剂ALA(中国奥立宝公司),链脲菌素(Streptozotocin,STZ;美国Sigma公司),总抗氧化物酶活性(total antioxidant capacity,T-AOC)、总超氧化物歧化酶活性(total superoxide dismutase,T-SOD)、过氧化氢酶活性(Catalase,CAT)和丙二醛(Malondialdehyde,MDA)试剂盒(南京建成公司),SnoN、TGF-β1多克隆抗体(美国Santa Cruz公司),Collagen I和Collagen IV单克隆抗体(美国Sigma公司),β-actin抗体(中国Boster公司),两步法免疫组织化学检测试剂盒、3-氨基-9-乙基卡唑(3-amino-9-ethylcarbozole,AEC)(中杉金桥生物技术有限公司),I抗稀释液,超敏显色液(enhanced chemiluminescent,ECL)、二喹啉甲酸(bicinchoninic acid,BCA)蛋白浓度测定试剂盒(碧云天生物技术研究所),Western blot用PVDF膜和3 mm Whatman滤纸(美国Millipore公司),BCA蛋白浓度测定试剂盒(碧云天生物技术研究所)。
1.1.3主要器材稳步倍加型血糖仪(美国强生公司),超低温冰箱(日本Sanyo公司),高速低温离心机(美国Beckman公司),Bayer1650全自动生化分析仪(美国Beckman公司),电泳系统及电转移装置(瑞典Amersham公司),凝胶成像系统(美国Bio-Rad公司)。
1.2 方法
1.2.1动物模型复制及分组SD大鼠适应性喂养一周后,尾静脉注射溶于0.01 mol/L(pH=4.5)无菌柠檬酸-柠檬酸钠缓冲液的STZ(55 mg/kg)复制DM大鼠模型,72 h后测大鼠空腹血糖,血糖≥16.7 mmol/L且尿糖阳性认为复制成功,随后随机分成糖尿病组(DM组,n =8)、硫辛酸治疗组(ALA 组,n =8)。成模 2周后,采取灌胃方式给予硫辛酸治疗,硫辛酸溶于5%的羧甲基纤维素钠(car-boxy methylated cellulose,CMC)中,灌胃剂量为150 mg/(kg·d),每周给药6 d,3 d配一次药(4℃保存);并设鼠龄相同的正常对照组(NC组,n =8),灌胃同等浓度同等剂量的CMC;6周后处死所有SD大鼠,期间所有大鼠予标准饲料喂养,自由饮水,每周监测1次血糖和体重。
1.2.2标本收集大鼠处死前1 d用代谢笼收集24 h尿,记录尿量,取部分尿液离心后-20℃保存;处死前禁食6~8 h,乙醚麻醉后称重,股动脉穿刺采血,分离血清-20℃保存;开腹取双侧肾脏,去掉包膜及周围脂肪组织,称重记录肾重/体重(kidney weight/body weight,KW/BW),分别用4%多聚甲醛固定及-80℃保存。
1.2.3生化指标测定葡萄糖氧化酶法测血清葡萄糖(blood glucose,BG),酶分析法检测血总胆固醇(total cholesterol,TC)、三酰甘油(Triglyceride,TG),邻苯三酚红比色法测尿蛋白(urine protein,UP),均按试剂盒说明书操作,尿蛋白浓度与尿量乘积为24 h UP。
1.2.4氧化应激水平检测每个肾组织标本均称取0.2 g按1∶9的比例与0.9%的生理盐水混合,置于匀浆器中冰上匀浆组织制成10%的组织匀浆,再用BCA蛋白浓度测定试剂盒测定组织匀浆的蛋白浓度。最后参照试剂盒说明书进行T-AOC、MDA、CAT和T-SOD的检测。其中,检测T-AOC和MDA时直接使用10%的组织匀浆;而检测CAT时将样本在10%的组织匀浆的基础上用生理盐水作40倍稀释,检测T-SOD时将样本作100倍稀释。4项指标的检测均采用比色法。
1.2.5肾组织病理检查多聚甲醛固定肾组织,制成3μm厚的石蜡切片,行HE及Masson染色,光镜观察肾组织形态结构变化。
1.2.6免疫组织化学染色免疫组织化学染色采用SP两步法检测Collagen I在各组大鼠肾组织的分布和表达,石蜡切片脱蜡水化,经3%的过氧化氢去离子水孵育及胰酶修复后,Collagen I(1∶100),4℃孵育过夜加入生物素化二抗,室温下孵育30 min,AEC显色,阳性染色为红色,苏木素复染,PBS代替一抗,作为阴性对照。
1.2.7Western blot检测取-80℃保存的各组大鼠肾皮质,每只样本取200 mg,分别加入组织蛋白提取液后匀浆离心取上清,用BCA试剂盒测定各组蛋白质浓度,按所测得浓度计算每泳道所需体积,加入加样缓冲液煮沸10 min,经8%的聚丙烯酰胺凝胶电泳分离,再转移至PVDF膜上,5%脱脂奶粉室温封闭1 h,分别加入 β-actin、SnoN、TGF-β1、Collagen Ⅳ一抗,工作浓度分别为1∶4 000、1∶300、1∶300、1∶1 000,4℃孵育12~24 h;次日,加入相应的辣根过氧化物酶标记的二抗(浓度均为1∶4 000)室温孵育1 h,加ECL荧光显色液,凝胶成像仪曝光,Image Lab软件分析各条带调整体积值,每个样本重复操作3次,以β-actin蛋白条带作为内参,结果用目标蛋白与β-actin的比值来表示。
1.3 统计学方法
采用SPSS 17.0软件进行统计学处理,实验数据以均数±标准差(±s)表示,若数据符合正态分布,通过方差齐性检验,多组比较采用单因素方差分析,组间两两比较采用LSD-t检验,P <0.05为差异有统计学意义。
2 结果
2.1 生化指标检测结果
大鼠在注射STZ 72 h后,BG升高并持续在高水平,且尿糖阳性。实验6周后,3组KW/BW、BG、24 h UP、TC和TG比较,采用单因素方差分析,差异有统计学意义(P <0.05),DM组的KW/BW、BG、24 h UP、TC和TG与NC组相比均升高(P <0.05);与DM组相比,ALA组KW/BW、24 h UP、TC和TG均降低(P <0.05),而BG增高,差异无统计学意义(P >0.05)。见表 1。
2.2 氧化应激水平检测结果
实验6周后,3组大鼠肾组织中T-AOC、MDA、T-SOD和CAT比较,采用单因素方差分析,差异有统计学意义(P <0.05);DM组T-AOC、T-SOD和CAT与NC组相比降低,使用ALA治疗后,ALA组T-AOC、T-SOD和CAT活性较DM组增高(P <0.05);而DM组MDA含量较NC组升高(P <0.05),ALA能降低DM大鼠肾组织MDA含量。见表2。
2.3 肾组织病理改变
HE及Masson染色可见正常大鼠肾小管结构清晰,肾小管上皮细胞排列整齐,基底膜完整,间质未见炎症细胞浸润;DM组大鼠肾小管腔扩张明显,肾小管基底膜不规则增厚,肾小管上皮细胞出现空泡状改变,间质出现炎症细胞浸润,肾小管间质Masson染色阳性物质增多;ALA组大鼠肾脏病变有不同程度的改善,肾小管间质Masson染色阳性物质减少,炎症细胞浸润减轻,空泡状改变不明显(见图1、2)。
2.4 免疫组织化学结果
免疫组织化学染色检测Collagen I的表达,NC组大鼠肾组织Collagen I阳性染色主要存在血管周围和细胞间质,而DM组大鼠阳性染色增多,可见肾小管基底膜强阳性染色,ALA治疗后 Collagen I的表达减少。见图3。
表1 各组大鼠KW/BW、BG、24 h UP、TC、TG的变化 (n =8,±s)
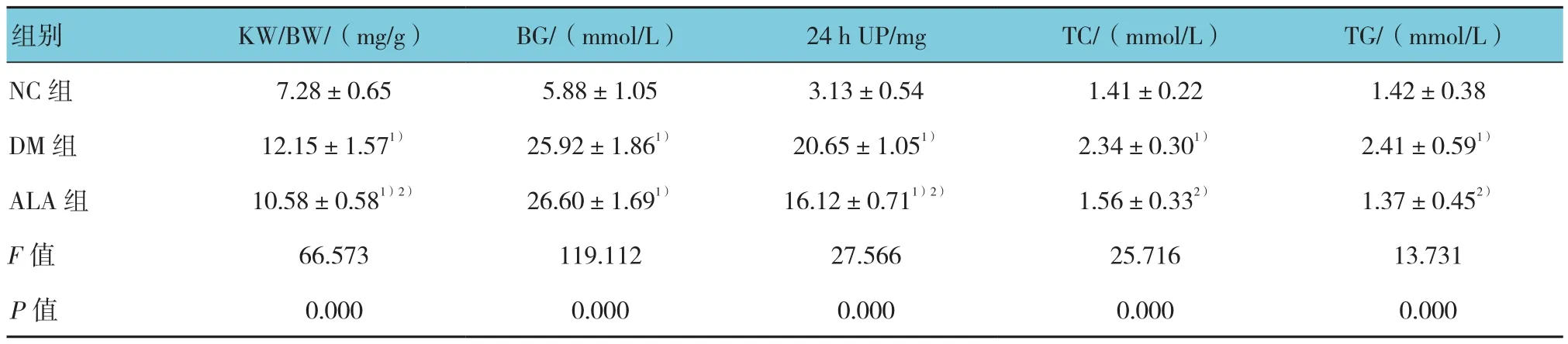
表1 各组大鼠KW/BW、BG、24 h UP、TC、TG的变化 (n =8,±s)
注:1)与NC组相比,P <0.05;2)与DM组相比,P <0.05
组别 KW/BW/(mg/g) BG/(mmol/L) 24 h UP/mg TC/(mmol/L) TG/(mmol/L)NC 组 7.28±0.65 5.88±1.05 3.13±0.54 1.41±0.22 1.42±0.38 DM组 12.15±1.571) 25.92±1.861) 20.65±1.051) 2.34±0.301) 2.41±0.591)ALA 组 10.58±0.581)2) 26.60±1.691) 16.12±0.711)2) 1.56±0.332) 1.37±0.452)F值 66.573 119.112 27.566 25.716 13.731 P值 0.000 0.000 0.000 0.000 0.000
表2 各组大鼠氧化应激水平的比较 (n =8,±s)
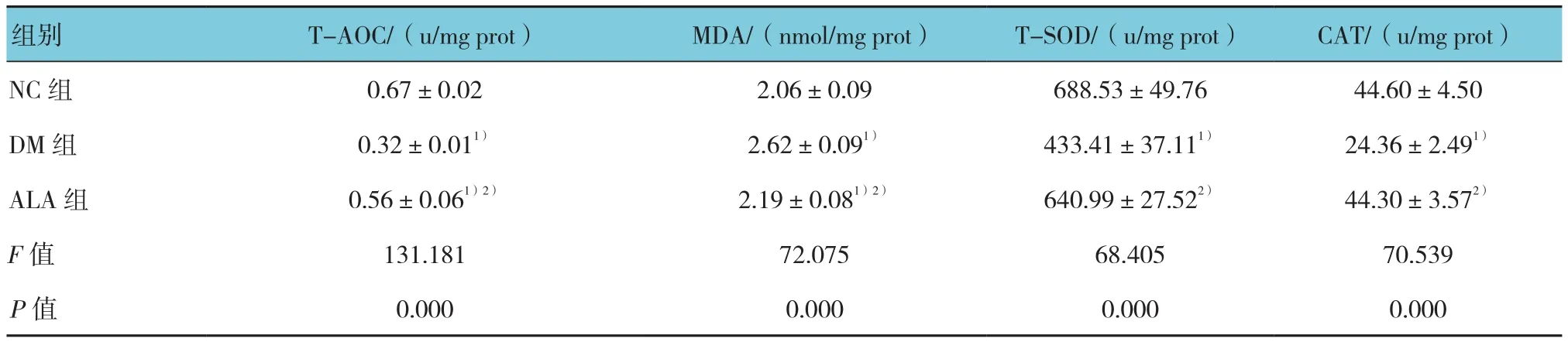
表2 各组大鼠氧化应激水平的比较 (n =8,±s)
注:1)与NC组相比,P <0.05;2)与DM组相比,P <0.05
NC 组 0.67±0.02 2.06±0.09 688.53±49.76 44.60±4.50 DM组 0.32±0.011) 2.62±0.091) 433.41±37.111) 24.36±2.491)ALA 组 0.56±0.061)2) 2.19±0.081)2) 640.99±27.522) 44.30±3.572)F值 131.181 72.075 68.405 70.539
2.5 Western blot结果
Western blot检测各组SnoN、TGF-β1和CollagenⅣ蛋白表达的水平,NC组中,SnoN条带较粗较强,而TGF-β1和CollagenⅣ条带较细较弱。与NC组相比,DM组中SnoN条带灰度值较少,TGF-β1和CollagenⅣ条带灰度值增加,差异有统计学意义(P <0.05);与DM组相比,ALA组SnoN条带变粗变强,TGF-β1和Collagen Ⅳ条带变细变弱,均差异有统计学意义(P <0.05),见图 4。
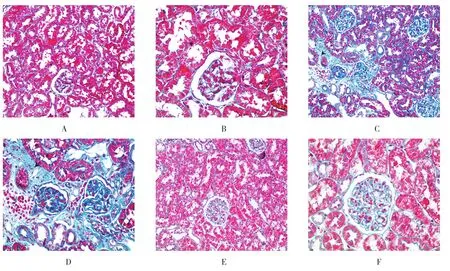
图2 各组大鼠肾组织形态 (Masson染色)
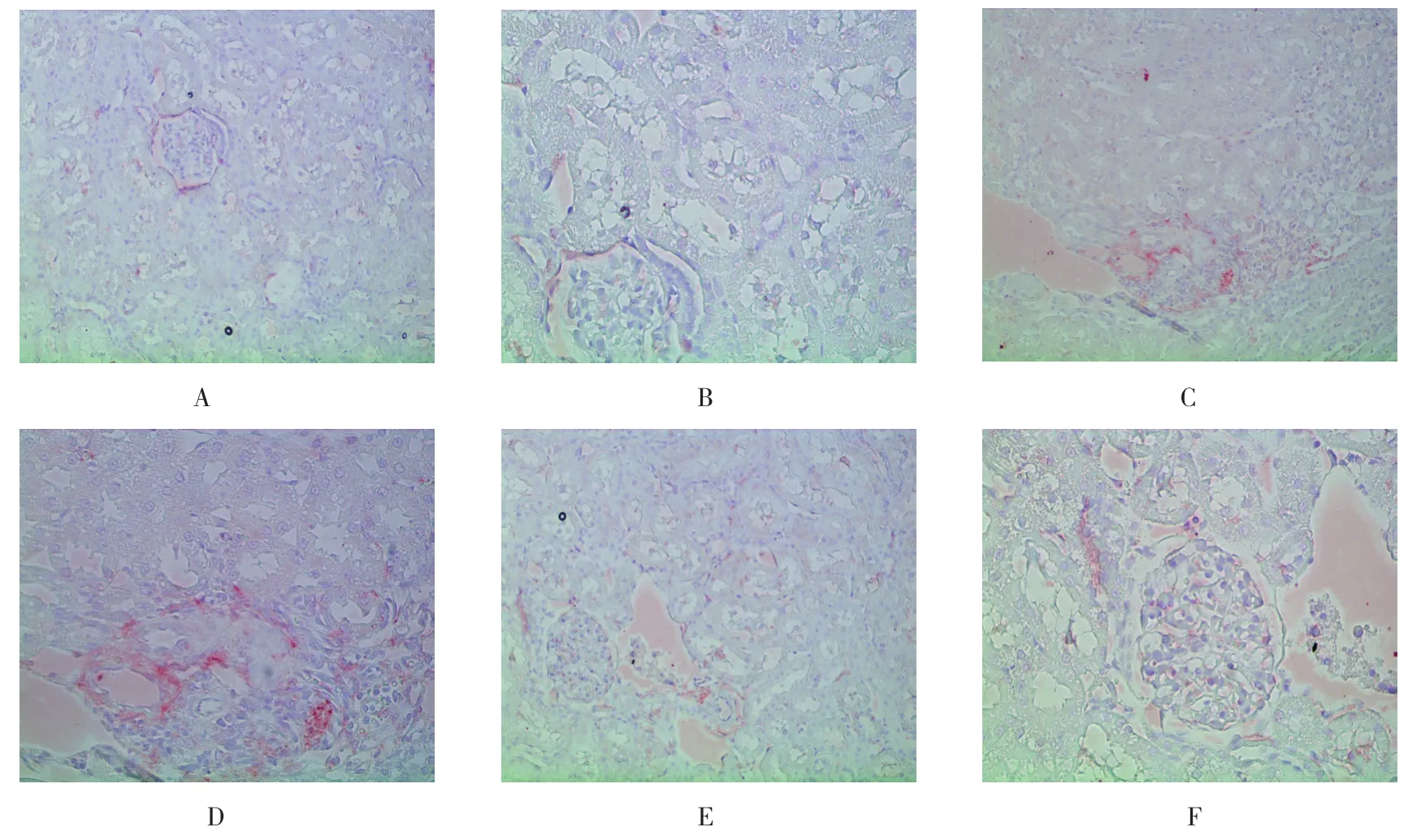
图3 各组大鼠肾组织Collagen I蛋白表达 (免疫组织化学染色)
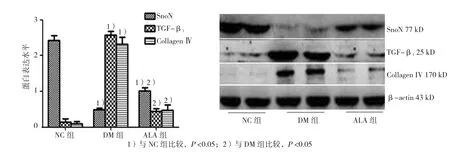
图4 各组大鼠肾组织中SnoN、TGF-β1和Collagen Ⅳ蛋白表达水平比较
3 讨论
氧化应激被认为是造成糖尿病及其并发症的重要原因之一[6-7]。ROS产生和清除之间的平衡决定了机体内氧化应激水平,ROS产生过多和/或抗氧化系统酶活性的降低可导致体内氧化应激水平增加。OS可以诱导肾脏发生损伤,主要表现为肾小球和肾小管固有细胞结构和功能上的改变[3]。有研究表明[8-9],肾脏中过多的OS可以破坏足细胞结构,使肾小球足细胞足突肥大;过多的ROS持续刺激还将诱导肾小管炎症反应和肾间质纤维化的形成。本研究复制DM大鼠模型6周后发现,DM大鼠肾组织脂质过氧化产物MDA含量升高而抗氧化物质T-AOC、T-SOD及CAT活性水平降低,且伴有明显的肾脏病理学改变,表明糖尿病时其氧化程度加重,抗氧化能力下降。而ALA作为一种抗氧化剂,可与其他抗氧化剂共同作用,抑制脂质过氧化[5,10]。本实验在DM大鼠模型复制2周后给予ALA治疗发现,ALA组血糖虽然没有明显变化,但病理学及生物化学检查显示ALA组肾脏病变程度减轻,同时在给予ALA治疗后大鼠肾脏脂质过氧化程度减轻,抗氧化能力增强。表明ALA可以降低DM大鼠肾脏的氧化应激水平,改善DM大鼠肾功能和代谢。
有研究表明[11],在糖尿病或高血糖状态下,肾脏内过多的ROS可激活胞内多条信号通路,包括MAPK通路、ERK通路等。还有研究表明[12],肾小管/间质内过多产生的ROS诱导TGF-β1等致纤维化因子大量释放和表达,促使肾间质纤维化的发生。说明肾脏内过多的ROS可能也诱导了TGF-β1信号通路的激活,从而加重其对肾脏的损害。而肾纤维化是DN终末期病理特征之一,有许多细胞因子参与这个过程,TGF-β1是目前公认最重要的致纤维化因子之一,主要通过介导TGF-β1信号通路来发挥其致纤维化效应[13-14]。SnoN作为TGF-β1信号通路的负调控因子,在肾脏纤维化过程发挥重要作用,它可以抑制TGF-β1信号通路的激活,并能抑制ECM沉积,从而达到延缓肾纤维化进程的目的[15-16]。本研究提示,DM组SnoN蛋白表达较NC组减少,TGF-β1水平与NC组比增高,并且伴随着Collagen I、Collagen Ⅳ的大量沉积;而用ALA治疗干预后,SnoN蛋白表达增多,TGF-β1蛋白表达和胶原沉积水平均下降。实验6周后,大鼠肾组织ROS产生增多,脂质过氧化程度加深,抗氧化能力减弱,促进TGF-β1高表达而抑制SnoN蛋白表达,导致SnoN调控TGF-β1信号通路的平衡被破坏,TGF-β1通过其下游胞内信号转导,促进目的基因Collagen I、Collagen Ⅳ的转录和翻译增多,致使Collagen I、Collagen Ⅳ在肾间质的沉积增多,DN发生纤维化病变;而用抗氧化剂ALA干预后DM大鼠肾脏脂质过氧化程度减轻,抗氧化能力增强,恢复SnoN蛋白水平并抑制TGF-β1表达,使两者之间的关系重新得到平衡,进而发挥延缓和治疗DN的作用。
综上所述,ALA减少了DM大鼠肾脏ROS的产生和脂质损伤,进而上调SnoN蛋白的表达、阻止TGF-β1信号通路的转导,从而发挥其延缓或阻断肾脏纤维化发生发展的作用,对DM大鼠肾脏起到保护作用。
参 考 文 献:
[1]SIES H. Biological redox systems and oxidatⅣe stress[J]. Cell Mol Li-fe Sci, 2007, 64(17): 2181.
[2]KASHIHARA N, HARUNA Y, KONDETI V K, et al. OxidatⅣe stress in di-abetic nephropathy[J]. Curr Med Chem, 2010, 17(34):4256.
[3]SINGH D K,WINOCOUR P, FARRINGTON K. OxidatⅣe stress in early dia-betic nephropathy: fueling the fi re[J]. Nat Rev Endocrinol, 2011, 7(3): 176.
[4]EDDY A A. Overview of the cellular and molecular basis of kidney fi brosis[J]. Kidney Int Suppl, 2014, 4(1): 2-8.
[5]BAST A, HAENEN G R. Lipoic acid. a multifunctional antioxidant[J]. Biofactors, 2003, 17: 207-213.
[6]PAN H Z, ZHANG L, GUO M Y. The oxidatⅣe stress status in diabetes me-llitus and diabetic nephropathy[J]. Acta Diabetol,2010, 47(suppl 1): 71-76.
[7]CERIELLO A. New insight on oxidatⅣe stress and diabetic complication may lead to a causal”antioxidant therapy[J]. Diabete Care, 2003, 26: 1589-1596.
[8]LEE H S. Mechanisms and consequences of TGF-overexpression by podocytes in progressⅣe podocyte disease[J]. Cell Tissue Res,2012, 347(1): 129.
[9]SINGH D K, WINOCOUR P, FARRINGTON K. OxidatⅣe stress in early diabetic nerphropathy: fueling the fi re[J]. Nat Rev Endocrinol, 2011, 7(3): 176.
[10]BHATTI F, MANKHEY R, MARIC C. Mechanisms of antioxidant and proo-xidant effects of alipoic acid in the diabetic and nondiabetic ki-dney[J]. Kidney Int, 2005, 67, 1371-1380.
[11]LEE H B, YU M R, YANG Y. ReactⅣe oxygen species regulated signali-ng pathways in diabetic nephropathy[J]. J Am Soc Nephrol, 2003, 14(8): S241.
[12]MASON R, WAHAB N. Extracellular matrix metabolism in diabetic ne-phropathy[J]. J Am Soc Nephrol, 2003, 14(5): 1358.
[13]VEGA G, ALARCON S, SAN MARTIN R. The cellular and signalling alter-ations conducted by TGF-β contributing to renal fi brosis[J]. Cytokine, 2016, 88: 115-125.
[14]KUMA A 1, TAMURA M, OTSUJI Y. Mechanism of and Therapy for Kidney F-ibrosis[J]. J UOEH, 2016, 38(1): 25-34.
[15]ZEGLINSKI M R, HNATOWICH M, DIXON I M. SnoN as a novel negatⅣ e regulator of TGF-β/Smad signaling:a target for tailoring organ fi brosis[J]. Am J Physiol Heart Circ Physiol, 2015,308(2): H75-82.
[16]LIU L, WANG Y, GUO B. Oxymatrine inhibits renal tubular EMT induced by high glucose via upregulation of SnoN and inhibition of TGFβ1/Smad signaling Pathway[J]. PLoS One, 2016, 11(3):e0151986.
猜你喜欢
传染病信息(2022年3期)2022-07-15
世界科学技术-中医药现代化(2022年2期)2022-05-25
上海交通大学学报(医学版)(2022年3期)2022-05-05
世界科学技术-中医药现代化(2020年2期)2020-07-25
健康之家(2020年15期)2020-05-08
中国医学创新(2019年9期)2019-08-19
中国中医急症(2019年10期)2019-05-21
中成药(2018年6期)2018-07-11
中成药(2017年12期)2018-01-19
医学研究杂志(2015年12期)2015-06-10
