
血管过氧化物酶1在Ang-Ⅱ诱导血管平滑肌细胞表型转化中的作用及机制
2018-03-22 06:17,,,,,
中南医学科学杂志 2018年1期
,, ,,,
(1.南华大学附属第一医院心血管内科,湖南 衡阳 421001;2.中南大学湘雅医院心血管内科)
血管平滑肌细胞(Vascular smooth muscle cells,VSMCs)表型转化即由收缩型向合成型转化在动脉粥样硬化、肺动脉高压、支架内再狭窄等血管增生性疾病中起重要作用[1-3],但调节VSMCs表型转化的具体机制仍不明确。研究表明氧化应激参与VSMCs表型转化[4],而VSMCs内的活性氧(Reactive oxygen species,ROS)主要来源于还原型烟酰胺腺嘌呤二核苷酸磷酸(Nicotinamide adenine dinucleotide phosphate,NADPH)氧化酶。血管过氧化物酶1(Vascular Peroxidase 1,VPO1)是一种新型的过氧化物酶,能催化NADPH氧化酶(Nicotinamide adenine dinucleotide phosphate oxidase,Nox)来源的过氧化氢(H2O2)产生氧化能力更强的次氯酸(Hypochlorous Acid,HOCl)加剧氧化应激[5]。血管紧张素Ⅱ(Ang-Ⅱ)通过上调Nox来源的ROS水平调节VSMCs的增殖和迁移[6],也有研究表明VPO1在Ang-Ⅱ诱导VSMCs增殖中起重要作用[7]。因此推测,Ang-Ⅱ上调VSMCs内VPO1的表达,并通过加剧氧化应激调节VSMCs表型转化。
1 材料与方法
1.1材料DMEM培养基购自美国Corning公司,胎牛血清、青霉素、链霉素、胰蛋白酶购自美国Gibco公司,兔VPO1抗体、兔α-SMA抗体、兔SM22α抗体、兔KLF4抗体、兔OPN抗体、兔GAPDH一抗、3-氯酪氨酸抗体均购自美国Abcam公司,BCA蛋白定量试剂盒、H2O2检测试剂盒、western blot 一抗稀释液均购自江苏碧云天生物有限公司,PVDF膜购自美国Millipore公司,VPO1siRNA、NC-siRNA购自中国广州锐博生物公司,逆转录试剂盒购自加拿大Fermentas公司,PCR引物购自中国上海程生物公司,Real-PCR试剂盒日本Takara公司。
1.2原代大鼠主动脉平滑肌细胞提取、培养选体重约为100~120 g雄性SD大鼠,用10%水合氯醛腹腔注射麻醉后置于75%乙醇溶液中浸泡3 min后处死。剪开胸腔,分离并剪开主动脉,剔除内皮细胞和血管外膜,将动脉中膜剪成约2 mm×2 mm大小的组织块均匀平铺于细胞培养瓶底壁,加入含20%胎牛血清、双抗(100 U/L青霉素+100 mg/L链霉素)、4.5 g/L葡萄糖的DMEM培养基,放置于37 ℃、5%CO2培养箱中,待细胞铺满瓶底面积约80%~90%后用清除组织块,予以0.25%胰蛋白酶消化传代。3~8代细胞用于细胞实验。
1.3免疫荧光鉴定血管平滑肌细胞将细胞培养于放有无菌盖玻片的6孔培养板内,待细胞密度生长约为60%,PBS润洗3次,每次10 min,4% 多聚甲醛固定15 min,PBS 漂洗3次后以0.1% Triton-X 100 室温处理20 min,5%BSA室温封闭30 min,加平滑肌肌动蛋白(smooth muscle α-actin,α-SMA)一抗(1∶1 000),4 ℃过夜后PBS 漂洗3次,每次10 min,加荧光二抗 (1∶1 000),避光孵育60 min,PBS漂洗后滴加DAPI(1∶10 000)5 min,PBS漂洗晾干后以10%甘油封片,避光保存,荧光显微镜下观察。
1.4 VPO1siRNA(si-VPO1)细胞转染接种约1×106个细胞至6孔板内,待每孔细胞密度生长约为30%~50%时,采用转染浓度为50 nmol/L siRNA严格按照试剂盒操作步骤进行转染,24 h后用real-time PCR和western blot检测转染效率。VPO1siRNA干扰序列为:5′-GUGGACUUGAAUGGAACAATT-3,由广州锐博生物公司合成,阴性对照siRNA(si-NC) 由该公司提供。
1.5 wstern blot蛋白检测提取细胞总蛋白,严格按照BCA蛋白定量试剂盒说明书测定蛋白浓度,取50 μg样本蛋白在10%SDS-PAGE的分离胶上进行蛋白电泳后转膜。将PVDF膜置于5%脱脂牛奶中室温下摇床上缓慢摇动封闭1 h,TBST漂洗3次,每次10 min,加入相应浓度的一抗孵育,4 ℃过夜后复温1 h,TBST漂洗3次,每次10 min,室温孵育二抗1 h,TBST漂洗3次,每次10 min。用凝胶图像处理系统(Bio-Rad)进行半定量分析。
1.6 Real-Time PCR mRNA检测使用Trizol(Invitrogen,USA)从培养的细胞中提取总RNA,并按照Prime ScriptTM RT Reagent Kit(TakaRa,Japan)试剂盒说明书逆转录成cDNA后,ABI 7500实时PCR系统(Foster City,USA)进行扩增。以GAPDH为内参对照,用ΔΔCt方法进行定量。引物序列如表1。
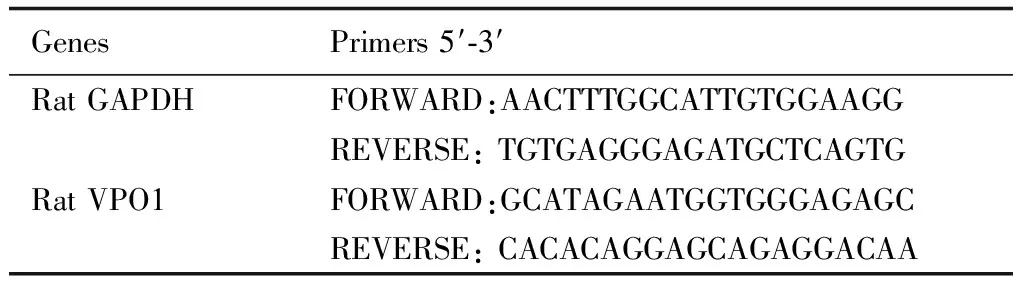
表1 引物序列
1.7 H2O2的检测收集细胞到离心管,加入适量的H2O2裂解液充分匀浆裂解细胞,4 ℃、12 000 rpm离心5 min,取上清做后续测定。H2O2检测试剂冰上溶解,标准溶液分别稀释成1、2、5、10、20、50 μmol/L,用96孔板检测,每孔加入50 μL标准品或者样品后,每孔再加入100 μL H2O2检测试剂,轻轻震荡混匀,室温静置30 min,立即测定A560的吸光值。
1.8 HOCl的检测因HOCl是一种氧化能力强且很不稳定的氧化剂,能与蛋白质酪氨酸残基发生氧化反应,生成稳定3-氯酪氨酸,因此通过western blot 检测3-氯酪氨酸表达情况可间接反应细胞内HOCl的生成[7]。
1.9统计方法所有数据均以均数±标准差表示,用SPSS 19.0软件进行分析。两组间比较检验正态性和方差齐性后采用t检验,多组间比较采用ANOVA分析。P<0.05为差异有统计学意义。
2 结 果
2.1原代VSMCs的鉴定α-SMA是VSMCs的特征性抗原之一,采用免疫荧光检测α-SMA鉴定VSMCs,结果显示提取的原代细胞中95%以上α-SMA免疫荧光染色呈阳性。见图1。
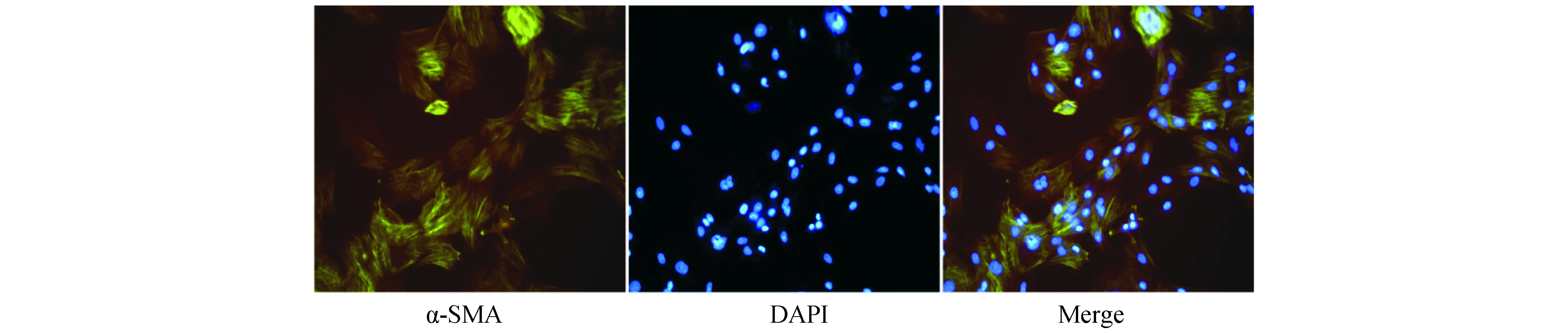
图1 免疫荧光鉴定VSMCs
2.2 Ang-Ⅱ对VSMCs内VPO1蛋白及mRNA表达的影响以不同浓度的Ang-Ⅱ(0 nmol/L、10 nmol /L、100 nmol/L、1 μmol/L)孵育VSMCs 24 h发现VSMCs内VPO1蛋白及mRNA的表达均呈浓度依赖性的升高。以终浓度为100 nmol/L的Ang-Ⅱ孵育VSMCs 0、12、24、48 h发现VSMCs内VPO1蛋白及mRNA均呈时间依赖性的升高。见图2。
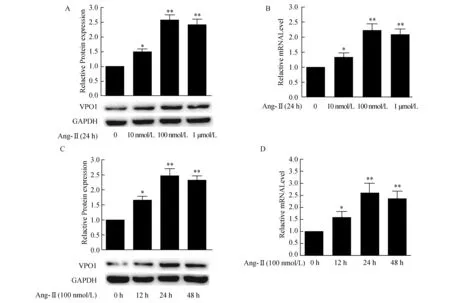
图2 Ang-Ⅱ对VSMCs内VPO1蛋白及mRNA表达的影响(n=4)A:western blot 检测VPO1蛋白表达的量效关系;B:RT-PCR检测VPO1mRNA表达的量效关系;C:western blot 检测VPO1蛋白表达的时效关系;D:RT-PCR检测VPO1mRNA表达的时效关系. 与Ang-Ⅱ0 nmol/L 0或0 h组比较,*P<0.05,**P<0.01
2.3 Ang-Ⅱ对VSMCs相关标记蛋白和KLF4蛋白表达的影响α-SMA、平滑肌22α(smooth muscle 22 alpha,SM22α)是VSMCs收缩型标记蛋白,骨桥蛋白(osteopontin,OPN)是VSMCs合成型的标记蛋白,Krüppel样因子4(KLF4)是调节VSMCs表型转化的关键转录因子,观察SM22α、α-SMA、OPN和KLF4蛋白的表达变化可评估VSMCs的表型转化。与对照组相比,以浓度为100 nmol/L Ang-Ⅱ孵育VSMCs 24 h后SM22α、α-SMA蛋白表达明显下降,而OPN、KLF4蛋白表达明显升高。见图3。
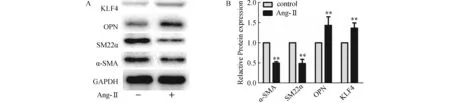
图3 Ang-Ⅱ对VSMCs相关标记蛋白和KLF4蛋白表达的影响(n=4)A:western blot 检测SM22α、α-SMA、OPN、KLF4蛋白的表达; B:SM22α、α-SMA、OPN、KLF4蛋白的相对表达. 与Control比较,**P<0.01
2.4 VPO1基因沉默的鉴定与空白转染组相比,转染si-VPO1组的VSMCs内VPO1 mRNA及蛋白表达明显下降。见图4。
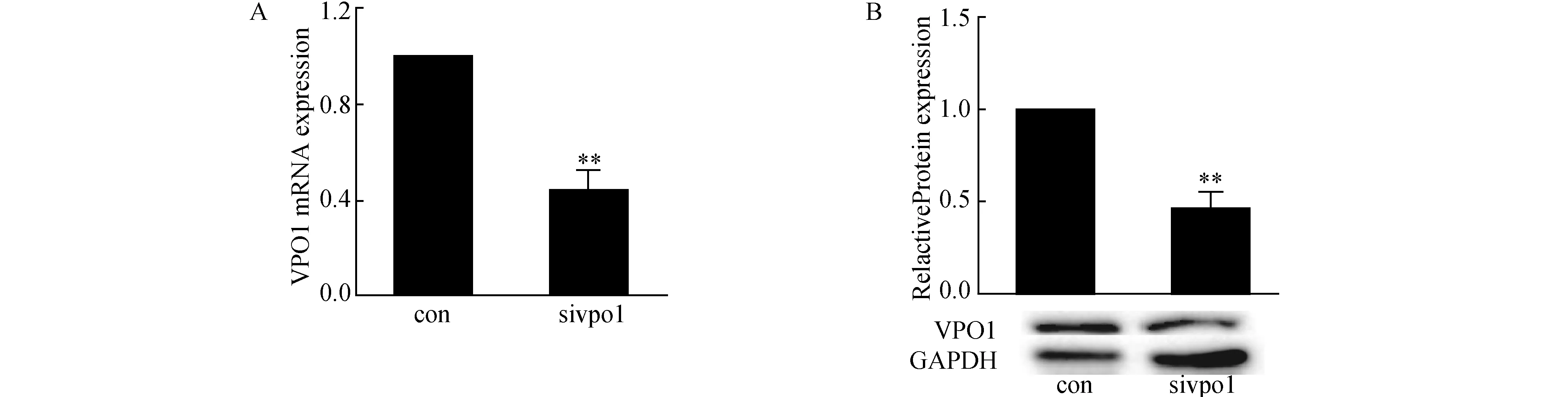
图4 VPO1基因沉默的鉴定(n=4)A:Real-time PCR检测VSMCs转染VPO1 siRNA后VPO1 mRNA的表达;B:Western Blot 检测VSMCs转染VPO1 siRNA后蛋白的表达. 与对照组相比,**P<0.01
2.5 VPO1基因沉默对Ang-Ⅱ培养VSMCs相关标记蛋白及KLF4蛋白表达的影响分别予以阴性对照siRNA (negative control siRNA) 或VPO1si RNA转染VSMCs 24 h,然后予以终浓度为100 nmol/L Ang-Ⅱ或不含Ang-Ⅱ的培养液VSMCs 24 h。结果显示:与si-NC组相比,Ang-Ⅱ+ si-NC组VSMCs内SM22α、α-SMA 蛋白减少,见图5。而OPN、KLF4蛋白表达增加;与Ang-Ⅱ+ si-NC组相比,Ang-Ⅱ+ si-VPO1组VSMCs内SM22α、α-SMA 蛋白表达增加,而OPN、KLF4蛋白表达减少。见图5。
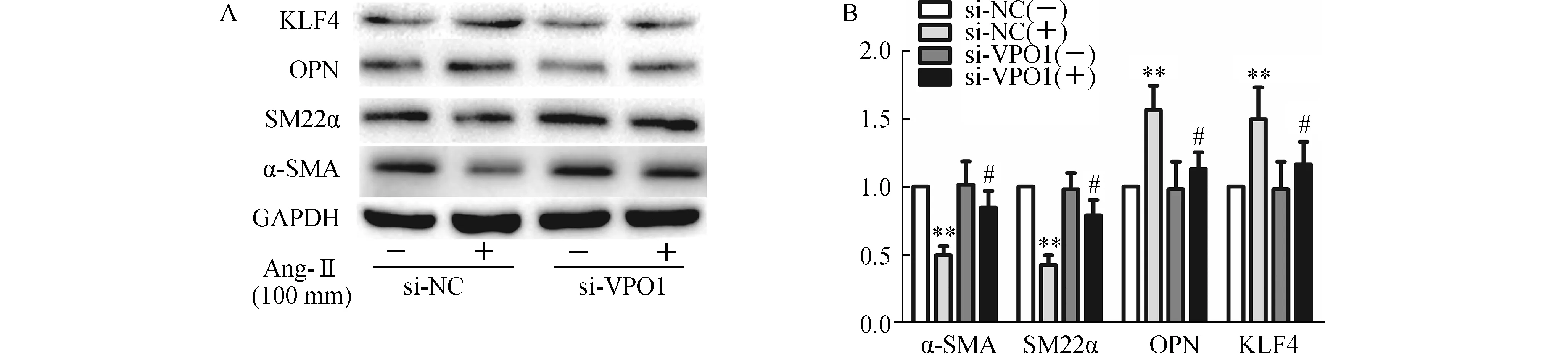
图5 VPO1基因沉默对Ang-Ⅱ培养VSMCs相关标记蛋白及KLF4蛋白表达的影响(n=4)A:Western blot检测α-SMA 、SM22α、OPN、KLF4蛋白表达;B:SM22α、α-SMA 、OPN、KFL4蛋白的相对表达.与NC-si RNA(-)组比较,**P<0.01;与NC-si(+)组比较,#P<0.05
2.6 VPO1基因沉默对Ang-Ⅱ培养VSMCs生成H2O2、HOCl的影响与si-NC组相比,Ang-Ⅱ+ si-NC组上清液中H2O2的生成增加;与Ang-Ⅱ+si-NC组相比,Ang-Ⅱ+ si-VPO组上清液中H2O2的生成减少,但无统计学意义(P>0.05)。与si-NC组相比,Ang-Ⅱ+ si-NC组3-氯络氨酸蛋白表达增加;与Ang-Ⅱ+si-NC组相比,Ang-Ⅱ+ si-VPO组3-氯络氨酸蛋白表达减少。见图6。
2.7 HOCl对VSMCs相关标记蛋白及KLF4蛋白表达的影响鉴于VPO1通过催化H2O2生成HOCl起作用,予以终浓度100 μmol/L HOCl培养VSMCs,结果发现:与对照组相比,HOCl组SM22α、α-SMA 蛋白表达减少,而OPN、KLF4蛋白表达增加。见图7。
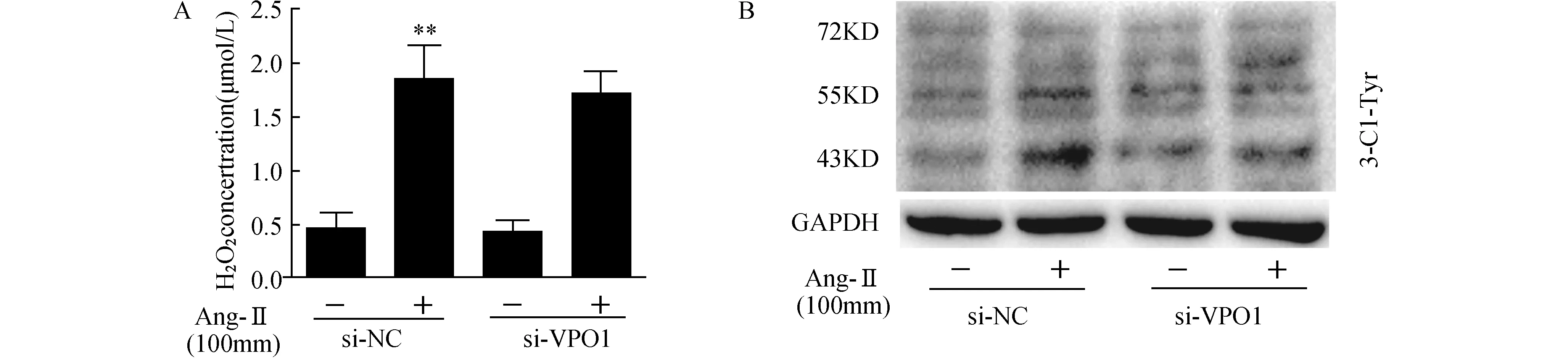
图6 VPO1基因沉默对Ang-Ⅱ培养VSMCs 生成H2O2、HOCl的影响(n=4)A:可见光光度法检测H2O2浓度;B:western blot检测3-氯络氨酸的表达间接反应细胞内HOCl的生成. 与NC-si RNA(-)组相比,**P<0.01
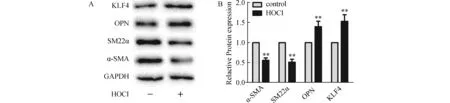
图7 HOCl对VSMCs相关标记蛋白及KLF4蛋白的影响(n=4)A:western blot 检测SM22α、α-SMA、OPN、KLF4蛋白的表达;B:SM22α、α-SMA、OPN、KLF4蛋白的相对表达. 与Control比较,**P<0.05
3 讨 论
VSMCs表型转化是其增殖、迁移及合成细胞外基质的前提,在动脉粥样硬化等血管增生性疾病中起重要作用[1]。最近研究发现VSMCs在人类新生内膜中大量聚集,被Allahverdian S 等[9]认为动脉粥样硬化是主要来源于VSMCs而非巨噬细胞参与的疾病。新近发表于Nature medicine研究发现在小鼠的动脉粥样硬化模型斑块内约80%的VSMCs收缩型标记蛋白α-SMA表达阴性,进一步证实VSMCs表型转化在动脉粥样硬化中作用[10]。肾素-血管紧张素系统(rennin-angiotensin system,RAS)在心血管疾病的病理生理过程中发挥重要作用,本课题予以Ang-Ⅱ培养VSMCs,结果发现Ang-Ⅱ下调VSMCs收缩型标志蛋白SM22α、α-SMA的表达和上调合成型标志蛋白OPN的表达,同时促进调节VSMCs表型转化的关键转录因子KLF4蛋白表达增加,表明Ang-Ⅱ诱导VSMCs表型转化。
氧化应激(oxidative stress)是指机体组织或细胞内氧自由基生成增加和(或)抗氧化应激能力降低,导致ROS在体内或细胞内蓄积而引起的氧化损伤过程。Rodriguez AI等[11]研究发现周期性拉伸的加强通过增加Nox1来源的ROS促进VSMCs由收缩型向合成型转化参与血管重构,表明Nox来源的ROS在VSMCs的表型转化中起重要作用。最近在VSMCs内发现的一种新型过氧化物酶即VPO1,类似于髓过氧化物酶(myeloperoxidase,MPO),能催化Nox来源的H2O2(弱氧化剂)产生HOCl(强氧化剂)加剧氧化应激损伤,在调节VSMCs增殖、成骨样转化及参与自发性高血压大鼠的血管重构中起重要作用[7,12,13]。本课题结果发现在Ang-Ⅱ诱导VSMCs由收缩型向合成型转化过程中,Ang-Ⅱ呈浓度和时间依赖性升高VSMCs内VPO1表达,伴随着H2O2和HOCl的生成增加。沉默VPO1基因后能明显抑制Ang-Ⅱ诱导VSMCs收缩型标记蛋白SM22α、α-SMA表达的减少和OPN表达的升高,伴随着细胞内HOCl生成减少。基于VPO1通过催化H2O2生成HOCl起作用,予以HOCl培养VSMCs,同样发现SM22α、α-SMA蛋白表达减少,而OPN蛋白表达增加,表明VPO1/HOCl途径参与VSMCs的表型转化。
KLF4作为锌指结构核转录因子 KLFs 家族重要成员之一,在VSMCs表型转化中起重要的调节作[14,15]。本课题予以Ang-Ⅱ培养VSMCs诱导其表型转化,结果发现Ang-Ⅱ升高VSMCs内VPO1表达的同时上调KLF4蛋白表达。沉默VPO1基因后明显抑制Ang-Ⅱ诱导VSMCs内KFL4蛋白的升高,表明VPO1通过调节KLF4的表达参与VSMCs的表型转化。同样我们予以HOCl培养VSMCs,发现细胞内KLF4蛋白表达明显增加。
总之,本课题在体外实验发现Ang-Ⅱ诱导VSMCs表型转化伴随VPO1蛋白的表达升高,并通过VPO1/HOCl途径调节KLF4蛋白的表达在Ang-Ⅱ诱导VSMCs表型转化中起重要作用。
[1] GOMEZ D,OWENS GK.Smooth muscle cell phenotypic switching in atherosclerosis[J].Cardiovasc Res,2012,95(2):156-64.
[2] ZHANG J,HU H,PALMA NL,et al.Hypoxia-induced endothelial CX3CL1 triggers lung smooth muscle cell phenotypic switching and proliferative expansion[J].Am J Physiol Lung Cell Mol Physiol,2012,303(10):L912-22.
[3] YAO Y,HU Z,YE J,et al.Targeting AGGF1 (angiogenic factor with G patch and FHA domains 1) for blocking neointimal formation after vascular injury[J].J Am Heart Assoc,2017,6(6):1-15.
[4] SUNAGA H,MATSUI H,ANJO S,et al.Elongation of long-chain fatty acid family member 6 (Elovl6)-driven fatty acid metabolism regulates vascular smooth muscle cell phenotype through AMP-activated protein kinase/kruppel-like factor 4 (AMPK/KLF4)signaling[J].J Am Heart Assoc,2016,5(12):1-17.
[5] LIU Z,LIU Y,XU Q,et al.Critical role of vascular peroxidase 1 in regulating endothelial nitric oxide synthase[J].Redox Biol,2017,12(1):226-32.
[6] JOJIMA T,UCHIDA K,AKIMOTO K,et al.Liraglutide,a GLP-1 receptor agonist,inhibits vascular smooth muscle cell proliferation by enhancing AMP-activated protein kinase and cell cycle regulation,and delays atherosclerosis in ApoE deficient mice[J].Atherosclerosis,2017,261(1):44-51.
[7] SHI R,HU C,YUAN Q,et al.Involvement of vascular peroxidase 1 in angiotensin II-induced vascular smooth muscle cell proliferation[J].Cardiovasc Res,2011,91(1):27-36.
[8] ZHANG H,XU H,Weihrauch D,et al.Inhibition of myeloperoxidase decreases vascular oxidative stress and increases vasodilatation in sickle cell disease mice[J].J Lipid Res,2013,54(11):3009-15.
[9] ALLAHVERDIAN S,CHEHROUDI AC,MCMANUS BM,et al.Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis[J].Circulation,2014,129(15):1551-9.
[10] SHANKMAN LS,GOMEZ D,CHEREPANOVA OA,et al.KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis[J].Nat Med,2015,21(6):628-37.
[11] RODRIGUEZ AI,CSANYI G,RANAYHOSSAINI DJ,et al.MEF2B-Nox1 signaling is critical for stretch-induced phenotypic modulation of vascular smooth muscle cells[J].Arterioscler Thromb Vasc Biol,2015,35(2):430-8.
[12] ZHUAN B,YU Y,YANG Z,et al.Mechanisms of oxidative stress effects of the NADPH oxidase-ROS-NF-kappaB transduction pathway and VPO1 on patients with chronic obstructive pulmonary disease combined with pulmonary hypertension[J].Eur Rev Med Pharmacol Sci,2017,21(15):3459-64.
[13] TANG Y,XU Q,PENG H,et al.The role of vascular peroxidase 1 in ox-LDL-induced vascular smooth muscle cell calcification[J].Atherosclerosis,2015,243(2):357-63.
[14] DING Y,ZHANG M,ZHANG W,et al.AMP-activated protein kinase alpha 2 deletion induces VSMC phenotypic switching and reduces features of atherosclerotic plaque stability[J].Circ Res,2016,119(6):718-30.
[15] WANG TM,CHEN KC,HSU PY,et al.microRNA let-7g suppresses PDGF-induced conversion of vascular smooth muscle cell into the synthetic phenotype[J].J Cell Mol Med,2017,21(12):3592-601.
猜你喜欢
昆明医科大学学报(2022年4期)2022-05-23
上海交通大学学报(医学版)(2022年3期)2022-05-05
湖南畜牧兽医(2021年6期)2022-01-24
河北果树(2021年4期)2021-12-02
食品安全导刊(2021年21期)2021-08-30
猪业科学(2021年5期)2021-06-02
天津医科大学学报(2021年1期)2021-01-26
医药前沿(2020年20期)2020-11-10
世界科学技术-中医药现代化(2020年2期)2020-07-25
质量安全与检验检测(2020年6期)2020-02-01
