
伴发闭角型青光眼视网膜色素变性遗传家系的表型分析和基因检测
2018-03-14 06:44:48李润璞刘铁城李淑贤陈晓菲代艾艾高旭辉
解放军医学院学报 2018年2期
李润璞,刘铁城,李淑贤,陈晓菲,代艾艾,高旭辉
解放军总医院 眼科,北京 100853
视网膜色素变性(retinitis pigmentosa,RP)是一组遗传性视网膜变性疾病,主要表现为从中周部向黄斑及中心凹蔓延的进行性感光细胞和色素上皮功能丧失[1-2],目前在全世界的发病率约为1/3 500[3],在中国约为1/3 467[4]。RP的遗传方式多样,非综合征型RP主要包括常染色体显性遗传(20% ~ 25%)、常染色体隐性遗传 (15% ~ 20%)、X-连锁遗传(10% ~ 15%)和散发型(30%),极少数是双基因型和线粒体相关型[1]。目前已经确认有83个致病基因与RP有关,其中可以表现为常染色体显性遗传的有28个[5]。然而除了典型的眼底表现外,有20% ~ 30%的患者伴随其他眼部症状甚至非眼部疾病,以Usher综合征(USH)和Bardet-Biedl综合征(BBS)最为常见。Usher综合征患者占所有RP患者的10% ~ 20%,最常见的伴随症状是不同程度的听觉障碍,部分伴有前庭性共济失调;占5% ~ 6%的Bardet-Biedl综合征患者除眼部症状外,还深受向心性肥胖、认知功能障碍、畸形、生殖腺发育不全和肾病的困扰[2]。同样的,伴发青光眼的急性发作或慢性进展也是RP患者视力急剧恶化的主要危险因素。本研究对一个来自中国山东的伴发闭角型青光眼的视网膜色素变性显性遗传家系进行临床诊断及基因检测,并对基因突变和临床表型的关系进行探讨。
资料和方法
1 对象 本研究符合《赫尔辛基宣言》原则。经我院伦理委员会批准,所有受检者均签署知情同意书。先证者来自山东省,2016年9月在本院眼科门诊诊断为视网膜色素变性,了解家族史后对其家系内6例成员进行检查,其中5例患者(Ⅲ:6、Ⅲ:9、Ⅳ:5、Ⅳ:9、Ⅳ:10),1例正常成员(Ⅲ:14)(图 1)。
2 临床资料 采集按照RP的诊断标准,询问家系成员病史,绘制家系图谱,门诊检查视力、眼压、视野、OCT等,裂隙灯及房角镜检查眼前节情况,散瞳后间接检眼镜查眼底并进行眼底照相,行视网膜电图(electroretinogram,ERG)检查。
3 外周血DNA制备 采集5例患者及1例正常成员的外周静脉血样品8 ~ 10 ml,乙二胺四乙酸(EDTA)抗凝。采用硅胶模技术及快速离心柱操作提取基因组DNA(QIAamp DNA Blood Midi Kit,Qiagen,Hilden,Germany)。紫外分光光度计及琼脂糖凝胶电泳检测DNA纯度合格,-80℃保存待用。
4 目标序列捕获与测序 使用Covaris LE220超声波仪(Massachusetts,USA)将先证者Ⅲ:9的DNA打断成200 ~ 250 bp的片段,然后Ampure Beads纯化,通过末端修复、加“A”以及加接头反应,制备先证者的DNA文库。使用定制RP基因片段捕获芯片(Roche NimbleGen,Madison,USA)与先证者DNA库杂交,随后进行芯片的洗涤、洗脱反应和已捕获样品的LM-PCR反应。使用Agilent 2100 Bioanalyzer 和ABI StepOne,进行文库的浓度、片段大小、富集度的检测,最后高通量测序仪Illumina HiSeq2500 Analyzers(Illumina,SanDiego,USA)连续双向测序90个循环并通过Illumina Pipeline software(version 1.3.4)读出原始测序数据[6]。
5 序列分析及验证 参考美国医学遗传学和基因组学学院(American College of Medical Genetics and Genomics,ACMG)相关指南解读数据,评估下机的原始数据(Raw reads)的测序质量,首先去除被接头污染以及低质量reads,随后用Burrows Wheeler Aligner软件[7]比对HG19序列,评价序列捕获效果,用SOAPsnp软件[8]和Samtools软件[9]分别进行SNV(single nucletide variant)和Indel(insertion and deletion)查询,生成目标区域碱基多态性结果,比对数据库NCBI dbSNP、HapMap、1000 Human Genome Dataset和 Database of 100 Chinese Healthy Adults,找出可疑突变,进行注释、筛选[6]。在所筛选出的致病突变所在片段的上下游设计引物并进行PCR扩增,对产物做Sanger测序,比对相关基因标准序列验证结果。使用同样的引物,对家系内其他成员的基因组DNA相关突变片段进行Sanger测序。
结 果
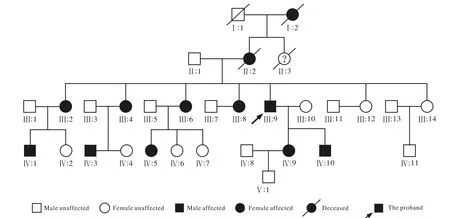
图1 视网膜色素变性家系系谱图Fig. 1 RP family tree chart
1 家系临床特征 先证者Ⅲ9的姐姐Ⅲ:6和子女Ⅳ:9、Ⅳ:10经检查均为患者,该家系RP性状连续4代传递,男女均有发病,比例为1∶2,符合常染色体完全显性遗传特征。5例患者,年龄28 ~ 60岁;夜盲为首发症状,平均发病年龄6岁,20岁之后出现不同程度视野缺损,平均41.5岁时视力明显下降,无色觉异常,Ⅲ:6、Ⅲ:9曾因长期眼痛、高眼压分别于7年前、3年前于外院行小梁切除+虹膜周切术,现眼压均在正常范围内(表 1)。
2 体征及检查 除Ⅳ:10外的4例患者均具有慢性闭角型青光眼相关的前节症状,裂隙灯下可见前房浅,晶状体表面散在少量色素,小梁可见不同程度色素沉积(图2A、图2B)。依照Scheie房角宽窄分类法,房角镜下患者房角呈窄Ⅰ~窄Ⅳ,先证者双眼均存在房角关闭(图2C、表2)。所有患者存在典型的RP表现:视网膜均呈青灰色,视盘颜色正常,杯盘比0.3,血管弓外可见轻度骨细胞样改变,此外Ⅲ:6、Ⅲ:9同时存在视网膜血管变细 (图 2D、图 2E)。Humphrey Zeiss 750i视野分析仪检测患者Ⅲ:9、Ⅳ:9、Ⅳ:10为上方视野受损,Ⅲ:6、Ⅳ:5为管状视野(图2F、图2G),经Zeiss Cirrus 5000 Hd-OCT检查可见除Ⅳ:10外的患者视网膜中心凹结构改变,中心凹外视网膜变薄,神经纤维层厚度减小(图2H、图2I、图2J)。使用Roland公司生产的视觉电生理检查系统(Ganzfeld Q450 ERG)检查见患者的a波、b波等均波幅重度降低或呈熄灭状态。Ⅲ:14检查无闭角型青光眼及RP表现。受检者眼底检查结果见表2。
3 测序结果及生物信息学分析 先证者目标序列捕获高通量测序共检测到突变89个,其中1个缺失突变和1个插入突变的突变位置均位于内含子,对氨基酸编码无影响,其余87个碱基置换突变中,有77个可以引起氨基酸编码顺序的改变。比较在互联网和本地数据库中的突变频率后,经生物信息学分析,9个<0.05的突变中有2个疑似致病(RHO基因c.541G>Ap.Glu181Lys突变和CNGA1基因的c.1271G>Ap.Arg424Gln突变)。
4 患者自身及家系内验证 对先证者及家系内其他成员的RHO基因c.541G>Ap.Glu181Lys突变和CNGA1基因的c.1271G>Ap.Arg424Gln突变进行Sanger测序,结果显示:RHO基因c.541G>Ap.Glu181Lys突变与临床表型共分离,验证结果见图3。
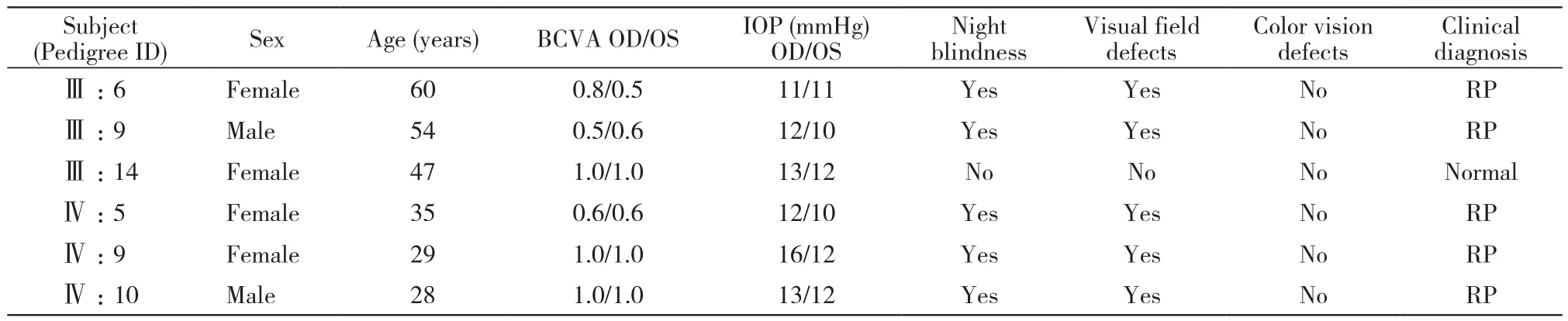
表l RP遗传家系的临床表型特点Tab. 1 Phenotypic characteristics of members in the RP family
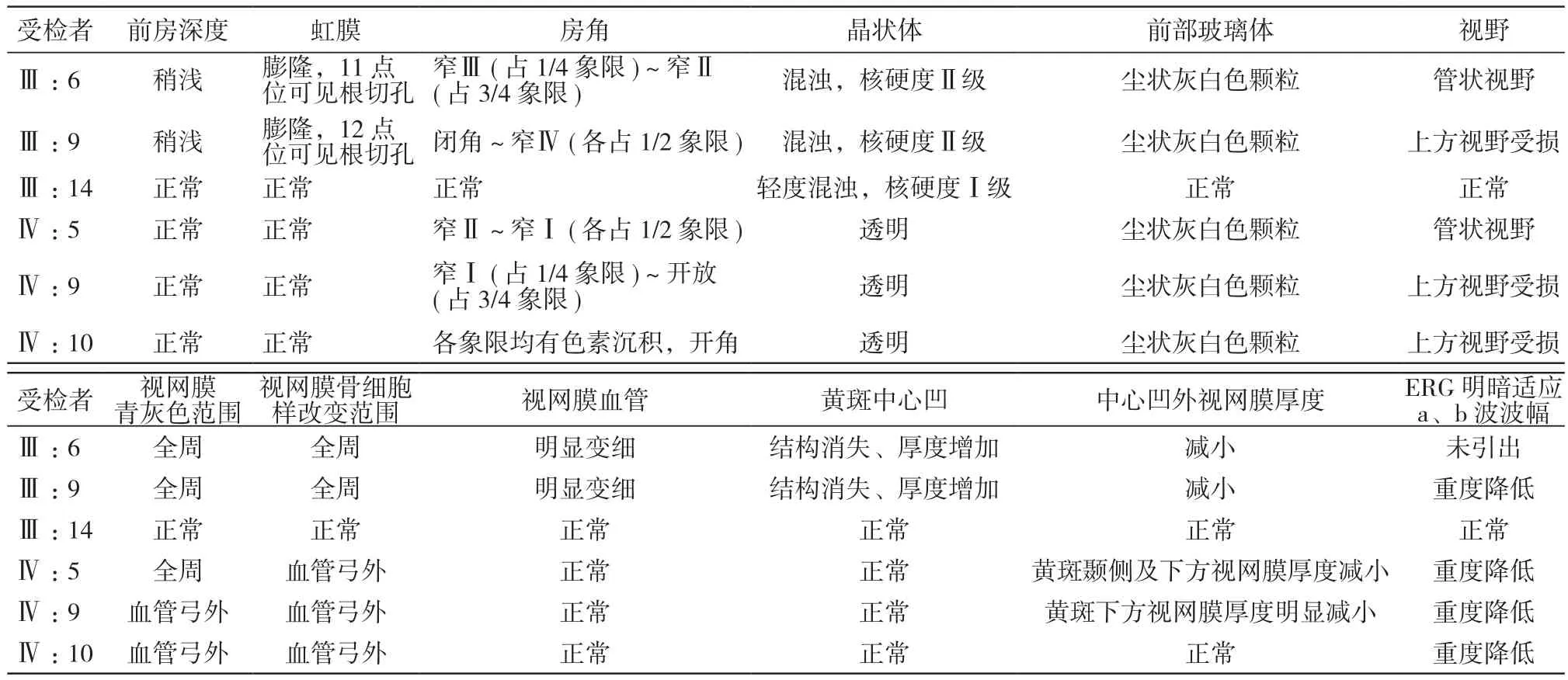
表2 RP患者检查结果Tab. 2 Results of ophthalmologic examinations in RP patients

图2 RP家系患者临床表型特点 A:患者Ⅲ:6右眼,可见11点位虹膜根切孔; B:患者Ⅲ:9右眼前房浅; C:患者Ⅲ:9右眼房角关闭;D:患者Ⅲ:6右眼视网膜全周青灰色、骨细胞样改变,视网膜血管明显变细; E:患者Ⅲ:9左眼视网膜骨细胞样色素沉积,下方血管瘤样改变; F:患者Ⅲ:9右眼上方视野受损; G:患者Ⅲ:6右眼管状视野; H:患者Ⅲ:9双眼视网膜纤维层厚度减小; I:患者Ⅲ:9右眼中心凹视网膜厚度增加; J:患者Ⅲ:6右眼黄斑外侧视网膜厚度明显减小Fig. 2 Clinical characteristics of RP patients A: patient Ⅲ 6, OD, showing an iris hole at 11 o'clock direction; B: patient Ⅲ 9,OD, showing shallow anterior chamber; C: patient Ⅲ 9, OD, showing chamber angle closure; D: patient Ⅲ 6, OD, showing attenuation of vessels, bone spicule deposits in the retina; E: patient Ⅲ 9, OS, showing bone spicule deposits in the retina,aneurysmal change of inferior vessels; F: patient Ⅲ 9, OD, showing superior visual field defect; G: patient Ⅲ 6, OD, showing a tubular visual field; H: patient Ⅲ 9, OD/OS, showing retinal nerve fiber layer thickness decrease; I: patient Ⅲ 9, OD, showing retinal thickness increase in central fovea; J: patient Ⅲ 6, OD, showing a thin retina beside macula lutea

图3 先证者Ⅲ:9 Sanger测序验证结果Fig. 3 Sanger sequencing results of the prob and Ⅲ:9
讨 论
根据本家系内患者的夜盲合并视野缺损病史、眼底表现和ERG特点,可以比较明确地做出RP的初步诊断。值得注意的是,患者Ⅲ:6、Ⅲ:9同时伴有闭角型青光眼,Ⅳ:5、Ⅳ:9也存在着不同程度的房角狭窄,提示RP患者可能存在青光眼易感性和家族聚集性。研究显示,RP患者伴发青光眼的发病率为2% ~ 12%[10]。根据彭清华[11]的统计分析,RP患者伴发青光眼的发病率是普通人群的2.13倍;Ko等[12]回顾分析382例台湾RP患者后提出,与正常人比较,RP患者伴发急性闭角型青光眼的概率是正常人的3.64倍。且在欧美开角型青光眼占多数,在我国闭角型青光眼较开角型少[10],考虑与种族差异有关。但RP患者伴发青光眼在临床上较易漏诊,主要原因是除急性发作型外多无疼痛等症状,且因RP的发病过程影响,视野改变不易诊断。本家系内Ⅲ:6、Ⅲ:9可见虹膜膨隆、浅前房、房角色素沉积,其轻微的视盘损害和正常范围杯盘比与长期高眼压病史不符,与典型闭角型青光眼不同,且视盘颜色变化较典型RP合并青光眼患者更加不明显,这一临床特点丰富了RP合并青光眼疾病的表型范围,更有助于以后的临床诊断。
目前,RP伴发青光眼的发病机制仍不明确,可能与原发性闭角型青光眼的病理过程不同[12],有报道认为原因在于变性的视网膜及视网膜色素上皮的毒性物质损害了小梁网的功能;从病理组织学来看,虹膜血管壁肥厚和葡萄膜血管壁异常与此有关;RP对其他器官及全身的影响,如黏多糖变性,也可导致了青光眼的发生[13]。本家系可能的致病原因:1)患者的原发性RP及年龄因素可能导致晶状体悬韧带退行性变化而松弛或稀疏,出现浅前房,“推挤”虹膜及睫状体导致继发性房角关闭[14];2)由于色素沉积在房水流出道,继而引起眼压上升,查阅文献虽未发现病理组织检查小梁网色素堵塞报告,但仍不排除为色素播散导致的复杂组织反应所致;3)从遗传学角度考虑,尽管RP和青光眼可以单独发病,但二者在基因的表达上有可能是相互影响的,这一理论仍需进一步研究来证实。
临床上为了防止漏诊,RP患者应常规进行眼压测量,必要时检查房角,以便早期诊断、早期治疗,延缓患者失明进程。此外,对于青光眼,尤其是闭角型青光眼,未能散瞳进行周边视网膜检查时,应调查患者有无夜盲及其RP家族史,从而判断有无合并RP对视野造成的影响。一旦确诊为RP合并青光眼,则建议患者接受家系调查,并进行遗传咨询。YAG激光周边虹膜成形术(LPI)的安全性、有效性及对视觉质量的较小影响[15],建议周边前房浅、房角镜检查确认房角窄的RP患者,尽早进行预防性LPI治疗。
本家系通过高通量测序平台,结合Sanger测序验证,初步认为致病原因与RHO基因EX3/CDS3上的c.541G>Ap.Glu181Lys杂合突变有关,此突变导致RHO蛋白181位的谷氨酸替换为赖氨酸,致病性有相关文献报道[16-18]。
人类RHO基因位于3q21-q24,长约6 952 bp,由5个外显子和4个内含子组成,编码的视紫红质(rhodopsin,RHO)蛋白是RP中发现的第一个突变蛋白。视紫红质是视杆细胞外节的视色素,包括一个由348个氨基酸组成的视蛋白和一个发色团(11-顺式视黄醛),被光子激发后发色团异构化为全反式视黄醛并导致光传导的激活,主要介导暗环境下的视力[19-20]。RHO基因是国内外已知的RP最常见致病基因之一[2,21-23],自Dryja等[24]在1990年首次报道adRP患者的RHO基因突变以来,迄今为止已经发现100多个与RP相关的RHO突变[25]。RHO基因不同的突变之间RP表型差异性较大,大多数情况下会导致常染色体显性遗传RP,但是也偶尔表现为常染色体隐性遗传RP[18]或者显性先天性静止性夜盲[26]。
本研究对一个伴发慢性闭角型青光眼的RP家系进行了临床检查和基因检测,丰富了RP合并青光眼疾病的表型范围,初步确认了致病基因突变。期望未来能结合更多的病例对RP合并青光眼的临床和基因诊断进行研究,进一步探讨两种疾病的内在关系,对临床诊疗提供指导。
1 Ferrari S, Di Iorio E, Barbaro V, et al. Retinitis pigmentosa: genes and disease mechanisms[J]. Curr Genomics, 2011, 12(4):238-249.
2 Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa[J].Lancet, 2006, 368(9549): 1795-1809.
3 Sullivan LS, Bowne SJ, Birch DG, et al. Prevalence of diseasecausing mutations in families with autosomal dominant retinitis pigmentosa : a screen of known genes in 200 families[J]. Invest Ophthalmol Vis Sci, 2006, 47(7): 3052-3064.
4 覃泳杰, 郭海科. 常染色体遗传型视网膜色素变性相关基因的研究进展[J]. 眼科研究, 2009, 27(12): 1159-1163.
5 RetNet[DB/OL]. https://sph.uth.edu/RetNet.
6 Wei X, Ju X, Yi X, et al. Identification of sequence variants in genetic disease-causing genes using targeted next-generation sequencing[J]. PLoS ONE, 2011, 6(12): e29500.
7 Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform[J]. Bioinformatics, 2009, 25(14):1754-1760.
8 Li R, Li Y, Fang X, et al. SNP detection for massively parallel whole-genome resequencing[J]. Genome Res, 2009, 19(6):1124-1132.
9 Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools[J]. Bioinformatics, 2009, 25(16): 2078-2079.
10 李美玉. 青光眼学[M]. 北京: 人民卫生出版社, 2004: 434-502.
11 彭清华. 视网膜色素变性并发青光眼21例临床分析[J]. 中国实用眼科杂志, 1995, 13(2): 90-91.
12 Ko YC, Liu CJ, Hwang DK, et al. Increased risk of acute angle closure in retinitis pigmentosa: a population-based case-control study[J]. PLoS ONE, 2014, 9(9): e107660.
13 王芳. 原发性视网膜色素变性伴发青光眼的研究进展[J]. 中华灾害救援医学, 2015, 3(8): 468-471.
14 Dikopf MS, Chow CC, Mieler WF, et al. Cataract extraction outcomes and the prevalence of zonular insufficiency in retinitis pigmentosa[J].Am J Ophthalmol, 2013, 156(1): 82-88.
15 Congdon N, Yan X, Friedman DS, et al. Visual symptoms and retinal straylight after laser peripheral iridotomy: the Zhongshan Angle-Closure Prevention Trial[J]. Ophthalmology, 2012, 119(7):1375-1382.
16 Xu Y, Guan L, Shen T, et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing[J]. Hum Genet, 2014, 133(10): 1255-1271.
17 Eisenberger T, Neuhaus C, Khan AO, et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis,non-coding exons and the overall variant load: the example of retinal dystrophies[J]. PLoS ONE, 2013, 8(11): e78496.
18 Dryja TP, Hahn LB, Cowley GS, et al. Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa[J]. Proc Natl Acad Sci USA, 1991, 88(20): 9370-9374.
19 Nathans J, Hogness DS. Isolation and nucleotide sequence of the gene encoding human rhodopsin[J]. Proc Natl Acad Sci USA, 1984, 81(15): 4851-4855.
20 Stojanovic A, Hwa J. Rhodopsin and retinitis pigmentosa: shedding light on structure and function[J]. Recept Channels, 2002, 8(1):33-50.
21 闫光辉, 盛迅伦. 中国视网膜色素变性相关基因研究现状[J].中华眼底病杂志, 2011, 27(5): 471-476.
22 Daiger S P. Perspective on Genes and Mutations Causing Retinitis Pigmentosa[J]. Archives of Ophthalmology, 2007, 125(2): 151.
23 Sullivan LS, Bowne SJ, Reeves MJ, et al. Prevalence of mutations in eyeGENE probands with a diagnosis of autosomal dominant retinitis pigmentosa[J]. Invest Ophthalmol Vis Sci, 2013, 54(9): 6255-6261.
24 Dryja TP, McGee TL, Hahn LB, et al. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa[J]. N Engl J Med, 1990, 323(19): 1302-1307.
25 Online Mendelian Inheritance in Man[DB/OL]. https://omim.org.
26 Dryja TP, Berson EL, Rao VR, et al. Heterozygous missense mutation in the rhodopsin gene as a cause of congenital stationary night blindness[J]. Nat Genet, 1993, 4(3): 280-283.
猜你喜欢
中国造纸(2022年9期)2022-11-25 02:24:54
临床输血与检验(2022年3期)2022-06-22 02:52:50
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
环球时报(2019-04-03)2019-04-03 04:15:14
幸福·健康版(2016年10期)2016-11-17 11:21:46
中国卫生标准管理(2015年2期)2016-01-14 03:41:36
西藏科技(2015年10期)2015-09-26 12:10:16
重庆医学(2015年12期)2015-03-05 05:52:54
中国当代医药(2015年1期)2015-03-01 02:00:17
