
水环境下蒙脱石层间CH4吸附行为的分子模拟研究
2018-02-05 08:24:35王建忠孙雨豪于欣畅刘熙远王浩王宣
西安石油大学学报(自然科学版) 2018年1期
王建忠,孙雨豪,于欣畅,刘熙远,王浩王宣
(中国石油大学(华东) 石油工程学院,山东 青岛 266580)
引 言
吸附态页岩气主要吸附于黏土矿物、有机质和干酪根上[1-2],而黏土矿物中,蒙脱石对页岩气的吸附能力最强[3]。蒙脱石是一种TOT型的膨胀性层状结构的黏土矿物[4],比表面积非常大[5],并且由于蒙脱石表面的原子移动困难,只能依靠吸附来降低自己的表面能,所以它是一种非常好的吸附剂。此外,页岩气储层中的蒙脱石一般含有层间水[6]。
国内外许多学者通过实验对页岩气的吸附规律进行了研究。例如,国外学者Lu X C等[7]研究发现,温度的升高会导致页岩气储层中的吸附气含量减少,游离气增多;Chalmers等[8]研究表明压力是影响页岩气吸附的主要原因之一,随着压力的增高,页岩气储层中的吸附气含量增大;国内学者张志英、杨胜波[9]实验研究了页岩气储层中的有机碳含量对页岩气吸附的影响,研究结果表明,页岩气的吸附量随着有机碳含量的增多而增多;吉利明等[3]对不同的黏土矿物进行了CH4吸附实验,对各种黏土矿物CH4吸附能力进行了排序:蒙脱石>>伊/蒙混石>高岭石>绿泥石>伊利石>粉砂石>石英砂岩。
室内的页岩气吸附实验研究可以直观地得到吸附规律,但是这些规律都过于宏观,很难发现页岩气吸附的微观机理。计算机分子模拟作为微观机理的研究方法,在石油行业中得到了广泛的应用。它不仅可以模拟常规实验难以达到的温度、压力条件,而且可以从微观上研究吸附机理。例如,Titiloye O[10]利用计算机分子模拟方法,从微观角度研究了CH4在蒙脱石层间的赋存以及运移机理;李文华[11]利用分子模拟研究了温度压力对CH4在蒙脱石层间吸附的影响,模拟规律与实验规律一致,并且得出CH4在蒙脱石层间的吸附属于物理吸附的结论;熊健等[12]利用分子模拟研究了CH4在蒙脱石狭缝孔中的吸附行为,而实验过程中难以观察纳米级狭缝的吸附情况。他的模拟表明:蒙脱石微孔中,CH4吸附量随着孔径增大而增大,而中孔中,CH4吸附量随着孔径增大而减小;隋宏光、姚军等[13-14]通过蒙特卡罗模拟研究了CH4和CO2在蒙脱石、干酪根中的竞争吸附,研究表明,CH4和CO2在蒙脱石狭缝中存在竞争吸附,蒙脱石的离子交换结构影响CO2的赋存状态,CH4和CO2在干酪根中的吸附符合langmuir吸附公式;Kadoura A,Nair A K N等[15]通过分子动力学的方法研究了CH4与CO2在蒙脱石层间的竞争吸附,研究表明,固定载荷的CO2在钠蒙脱石层间的扩散对CH4的动力学行为影响不大。
页岩气吸附实验和分子模拟的研究重点放在了温度、压力和有机碳含量对页岩气吸附量的影响上。但是地层环境复杂,这几种因素并不能全面解释页岩气吸附变化规律。考虑到计算机分子模拟所具有的优势,本文采用分子模拟的方式,将水环境作为一种影响因素,研究蒙脱石层间CH4的吸附行为。本文首先构建了钠蒙脱石的层间结构模型,使用分子力学的方法对模型进行优化,然后使用蒙特卡罗方法分别进行了H2O和CH4的吸附模拟,以及H2O和CH4的竞争吸附模拟。最后对蒙特卡罗方法模拟出来的结果进行分子动力学模拟,研究H2O和CH4在蒙脱石层间的吸附扩散行为。
1 模拟方法
1.1 位能模型的选取
有关蒙脱石的分子模拟一般采用表1的电荷数据以及L-J势能参数[16-19],运行结果较好。本文模拟直接引用表1的电荷数据、L-J势能参数。水分子模型采用常见的SPC/E模型,势能[16-17]
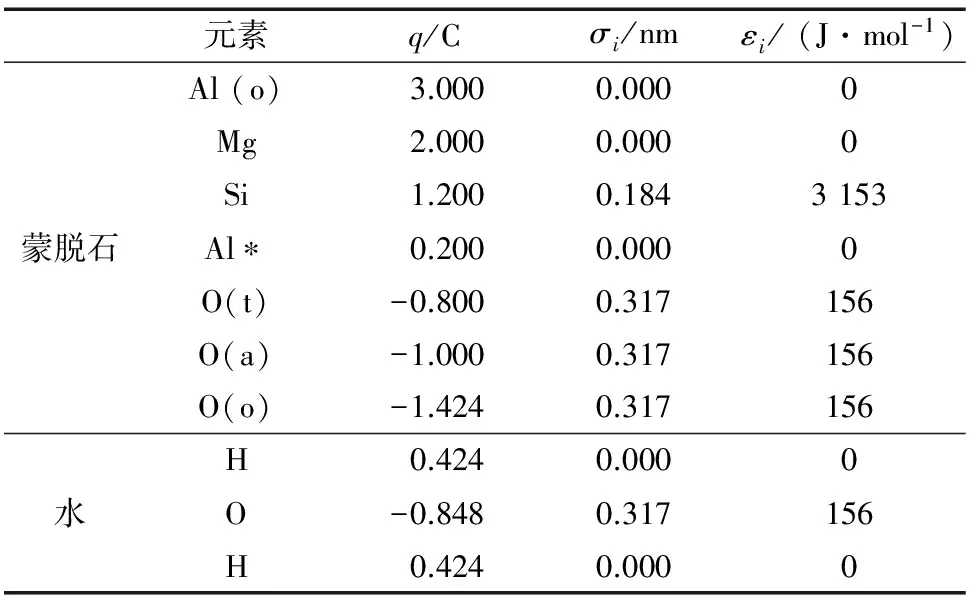
表1 蒙脱石、SPC/E水中各元素的电荷q和Lennard-Jones参数σi,εiTab.1 Charge q and Lennard-Jones parameters σi,εi of every elements in montmorillonite and SPC/E water
(1)
式中:Vij为两原子之间的势能,J ;qi、qj为原子电荷,C ;ε0为介电常数,F/m ;σij为L-J作用的尺度参数,m;εij为L-J作用的能量参数,J·mol-1;rij为两原子之间的距离,m。
式(1)中的相互作用常数σij,εij可根据Lorentz-Berthelot定律[16-19]
(2)
求得。式中:σi、εi和σj、εj分别为i、j原子的Lennard-Jones参数。
1.2 模型的建立
本文使用Materials Studio软件构建了钠蒙脱石的层间结构,使用的晶格参数见表2。最终所构建的蒙脱石模型如图1所示,分子式为:Na0.75·nH2O[Si7.75Al0.25][Al3.5Mg0.5]O20(OH)4。

表2 蒙脱石晶格参数Tab.2 Lattice parameters of montmorillonite
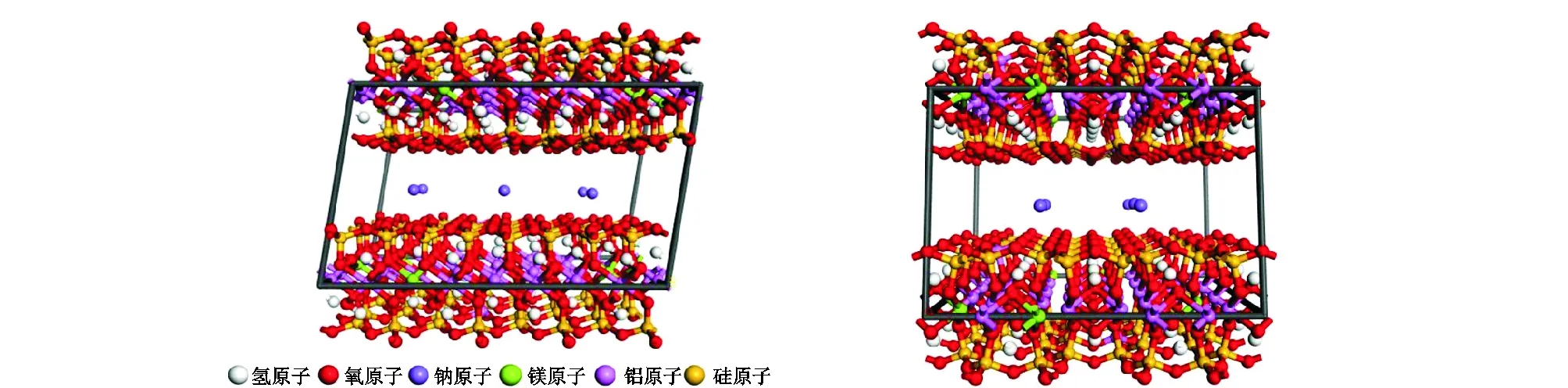
图1 蒙脱石结构透视效果Fig.1 Perspective view of montmorillonite structure
2 H2O/ CH4单组分吸附等温线
通过巨正则系综蒙特卡罗方法可以获得等温吸附曲线。蒙特卡罗方法[11]是一种可用于计算指定系综下气体吸附量、吸附热、吸附等温线的方法。计算时采用统计力学的方法对体系中的粒子进行位移、旋转以及粒子在相与相之间的转移。Materials Studio软件中采用Metropolis方法取样,对体系进行一系列取样之后的微观粒子逐渐趋于Boltzmann分布。
图2是H2O的等温吸附曲线,当压力在0.01~1 MPa之间递增时,H2O的吸附量剧烈增加,这一现象符合蒙脱石极易吸附H2O的特性[20];在压力增大到1 MPa之后,吸附量增加的速率变缓,最终吸附达到饱和,吸附量不再增加。随着温度的增加,H2O的吸附量减少。图3 是CH4的等温吸附曲线,温度与压力对CH4的吸附影响与其对H2O的影响规律一致。不同点在于,H2O的吸附量远大于CH4在蒙脱石层间的吸附量。可以通过吸附热来分析造成吸附量差异的原因:模拟吸附平衡之后,H2O的平均吸附热为70.20 kJ/mol,大于42 kJ/mol,说明有化学吸附的存在。CH4的平均吸附热是40.80 kJ/mol,低于42 kJ/mol,说明以物理吸附为主。
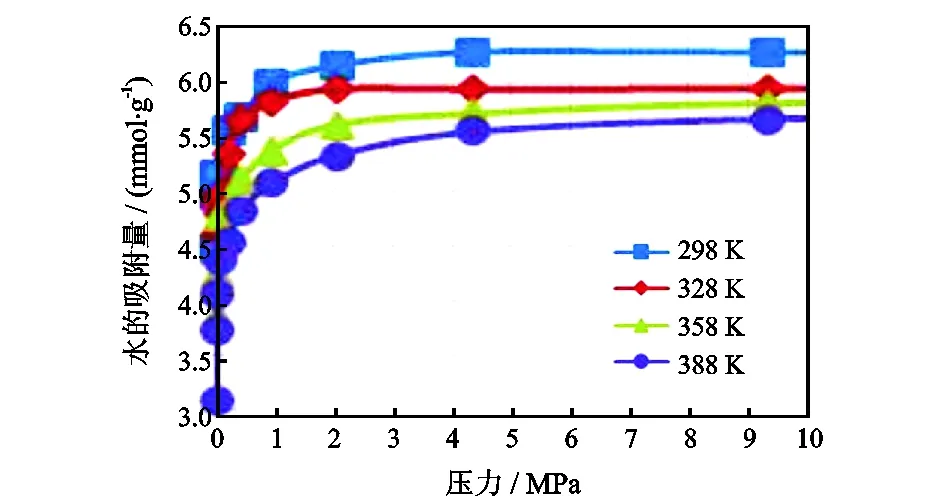
图2 H2O的等温吸附曲线Fig.2 Isothermal adsorption curve of H2O

图3 CH4的等温吸附曲线Fig.3 Isothermal adsorption curve of CH4
3CH4、H2O在蒙脱石层间的竞争吸附
图4是CH4和H2O在蒙脱石层间的竞争等温吸附曲线。平衡吸附时,CH4的吸附量维持在0.5 mmol/g左右,H2O的吸附量维持在5.1 mmol/g左右。从分子直径上看,H2O的直径是0.40 nm,CH4的直径是0.38 nm,CH4的直径略小于H2O的直径,在占据吸附位的行为上CH4应该占有微小的优势,但是从模拟的结果上看,对比单组分平衡吸附时的吸附量(对比图2、图3),CH4的吸附量减少了84.8%,而H2O的吸附量只减少了12.3%。因此,CH4和H2O在蒙脱石层间发生了明显的竞争吸附。
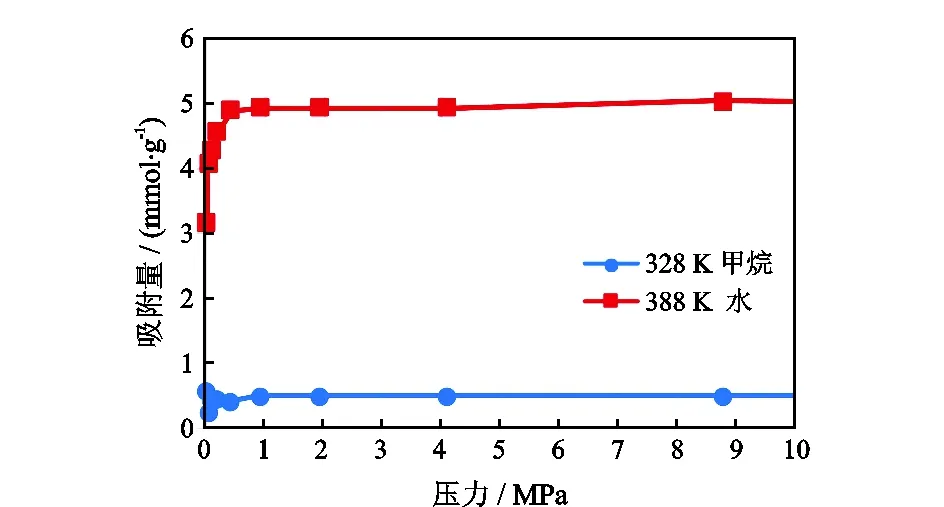
图4 CH4、H2O竞争等温吸附曲线Fig.4 Competitive isothermal adsorption curves of CH4 and H2O
4 层间水含量对CH4吸附的影响
4.1 不同水含量下CH4的等温吸附曲线
图5是温度为328 K,不同层间水含量下CH4的等温吸附曲线。随着层间水含量的增大,CH4的吸附量逐渐减少。但是不论层间水含量如何变化,CH4的吸附热却维持在40 kJ/mol左右,说明层间水的存在并不影响CH4的吸附能力,说明造成CH4吸附量减少的主要原因是H2O占据了CH4分子的吸附位。
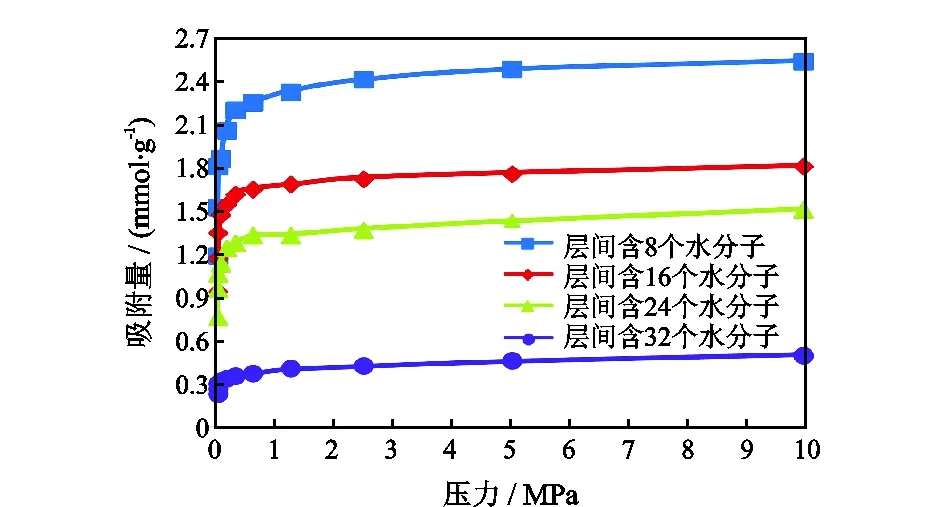
图5 不同层间水含量CH4的等温吸附曲线Fig.5 Isotherm adsorption curve of CH4 under different interlayer water content
4.2 不同层间水含量下的分子动力学模拟
在软件sorption模块中通过设置任务Fixed Loading,分别设置蒙脱石层间H2O个数为8、16、32、40,不断加大CH4的吸附个数,直到CH4吸附达到饱和为止。使用这几个最终的平衡吸附构型进行分子动力学模拟,采用NVT系综,设置温度为328 K,模拟时间为1 000 ps。分子动力学模拟[21]是一门多学科交叉的计算机模拟技术,方法主要依靠牛顿力学来模拟分子的运动。体系内各个原子坐标势能的总和构成了分子的内势能
Fi=-iV=-i(i+j+k)V。
(3)
在模拟开始的时候,首先给分子一个随机的速度,运用牛顿定理
(4)
(5)
vi=vi0+ait,
(6)
(7)
对牛顿运动方程中的时间积分,就可以模拟体系中间原子或分子在t时间时的运动速度和位置,根据t时间段内的运行情况,可以分析体系的物理性质变化以及结构变化特征。模拟的时间越长,结果越精确。
通过观察分子动力学模拟的整个过程可以发现,绝大部分CH4都是处于游离态,而层间H2O存在3种形态(如图6所示):(a)游离水;(b)氢键结合的表面水;(c)与金属阳离子结合的水。
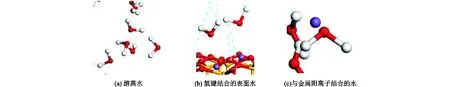
图6 水在蒙脱石层间存在的3种形态Fig.6 Three forms of water existing between montmorillonite layers
图7为不同水含量在运行动力学模拟之后001剖面上H2O的相对浓度分布图,可以看出H2O含量较少的时候,在两侧壁面分别形成一个吸附层。H2O含量较多的时候,水分子在壁面附近的吸附达到饱和,剩下的H2O除了与金属阳离子形成结合水以外,还有一部分在蒙脱石中央呈现出游离态,所以呈现出3个波峰。但是左右两侧的吸附量并不均匀,观察可知,层间Na+的分布也并不均匀,而H2O会与层间金属阳离子形成结合水,所以可能导致吸附层左右两侧的吸附量不平衡。
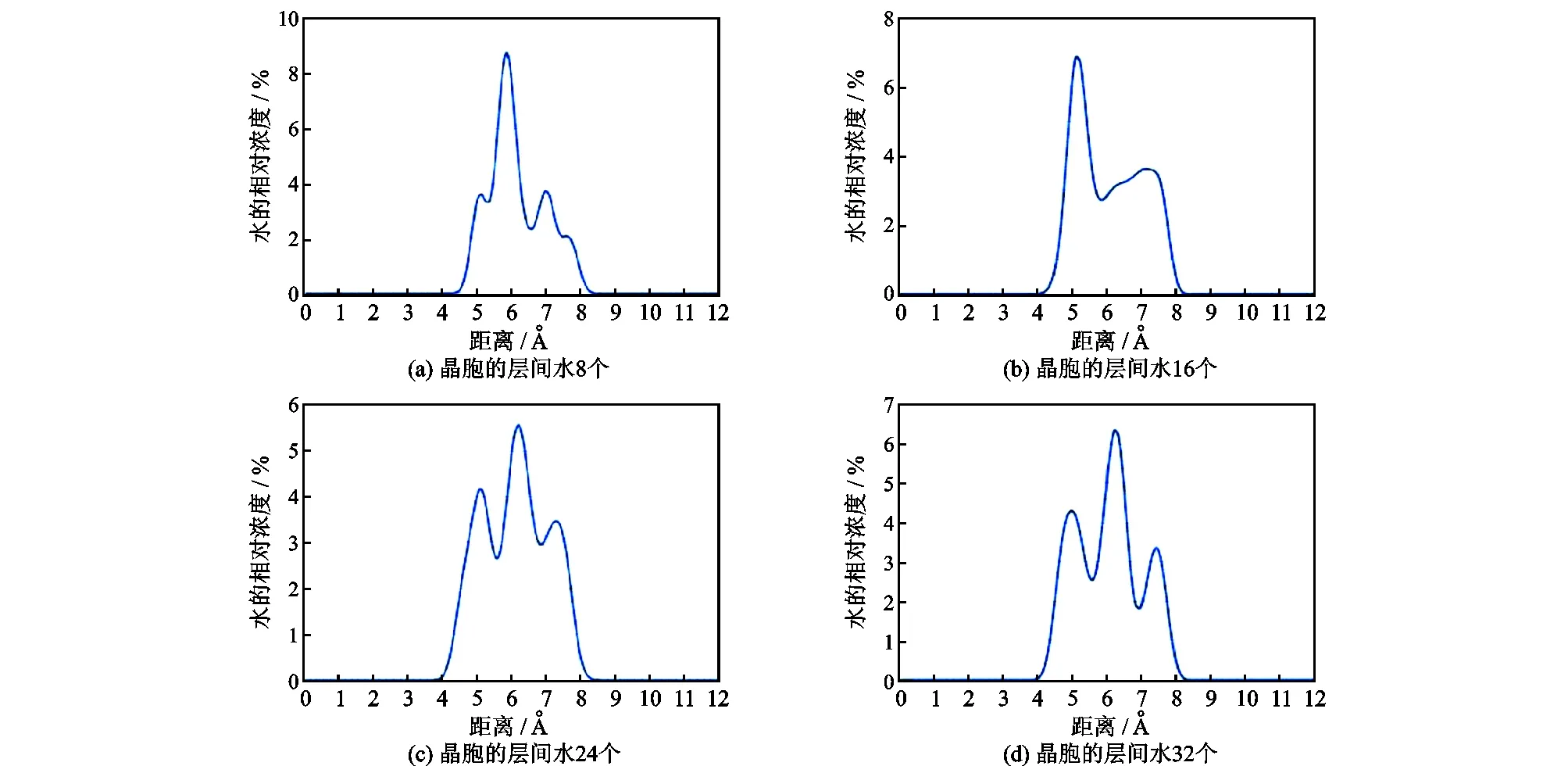
图7 H2O相对浓度分布Fig.7 Relative concentration profiles of H2O
图8是在不同含水量条件下,进行分子动力学模拟之后得到的0 0 1剖面上CH4的相对浓度分布图。对比图7可以发现,蒙脱石结构壁面优先吸附的是H2O,然后才开始吸附CH4。在观察分子动力学模拟的全过程中,CH4在蒙脱石壁面上停留的时间很短,可以认为是吸附与解吸同时发生,大部分的CH4处于游离态。

图8 CH4相对浓度分布Fig.8 Relative concentration profiles of CH4
5 结 论
(1)随着温度的增加,H2O和CH4在蒙脱石层间的吸附量减少。随着压力的增大,H2O和CH4在蒙脱石层间的吸附量逐渐增大。
(2)H2O与CH4存在竞争吸附,与单组分吸附相比,竞争吸附情况下,H2O的吸附量下降12.3%,CH4的吸附量下降了84.8%,竞争吸附明显。H2O在蒙脱石层间的吸附热大约为70.20 kJ/mol,属于化学吸附。CH4在蒙脱石的层间吸附热大约为40.80 kJ/mol,主要属于物理吸附。所以蒙脱石会优先吸附H2O。
(3)层间水的存在并不影响CH4的吸附能力,而造成CH4吸附量减少的主要原因是H2O占据了CH4分子的吸附位。通过分子动力学模拟观察可知,CH4在蒙脱石层间处于游离态,而水在蒙脱石层间的状态有:游离水、氢键结合的表面水以及与金属阳离子结合的水。
[1] 姚军,孙海,黄朝琴,等.页岩气藏开发中的关键力学问题[J].中国科学:物理学 力学 天文学,2013,43(12):1527-1547.
YAO Jun,SUN Hai,HUANG Chaoqin,et al.Key mechanical problems in the development of shale gas reservoirs[J].Scientia Sinica(Physica,Mechanica & Astronomica),2013,43(12):1527-1547.
[2] 董丙响,程远方,刘钰川,等.页岩气储层岩石物理性质[J].西安石油大学学报(自然科学版),2013,28(1):25-28,36.
DONG Bingxiang,CHENG Yuanfang,LIU Yuchuan,et al.Reserch of the petrophysical property of shale gas reservoirs[J].Journal of Xi'an Shiyou University(Natural Science Edition),2013,28(1):25-28,36.
[3] 吉利明,邱军利,张同伟,等.泥页岩主要黏土矿物组分甲烷吸附实验[J].地球科学(中国地质大学学报),2012,37(5):1043-1050.
JI Liming,QIU Junli,ZHANG Tongwei,et al.Experiments on methane adsorption of common clay minerals in shale[J].Earth Science(Journal of China University of Geosciences),2012,37(5):1043-1050.
[4] 高海英,杨仁斌,龚道新.蒙脱石的吸附行为及其环境意义[J].农业环境科学学报,2006,25(s1):438-442.
GAO Haiying,YANG Renbin,GONG Daoxin.Adsorption behavior of microorganisms and environmental significant[J].Journal of Agro-environment Science,2006,25(s1):438-442.
[5] VOLZONE C.Selective gas adsorption by amorphous clay-mineral derivatives[J].Clays & Clay Minerals,1999,47(5):647-657.
[6] 王行信.蒙脱石的成岩演变与石油的初次运移[J].沉积学报,1985,3(1):81-91.
WANG Xingxin.Diagenetic evolution of montmorillonite and primary migration of petroleum[J].Acta Sedimentologica Sinica,1985,3(1):81-91.
[7] LU X C,LI F C,WATSON A T.Adsorption measurements in Devonian shales[J].Fuel,1995,74(4):599-603.
[8] CHALMERS G R,BUSTIN R M.Lower Cretaceous gas shales in northeastern British Columbia,Part I:geological controls on methane sorption capacity[J].Bulletin of Canadian Petroleum Geology,2008,56(1):1-21.
[9] 张志英,杨盛波.页岩气吸附解吸规律研究[J].实验力学,2012,27(4):492-497.
ZHANG Zhiying,YANG Chengbo.On the adsorption and desorption trend of shale gas[J].Journal of Experimental Mechanics,2012,27(4):492-497.
[10] TITILOYE J O,SKIPPER N T.Monte Carlo and molecular dynamics simulations of methane in Potassium montmorillonite clay hydrates at elevated pressures and temperatures[J].Journal of Colloid and Interface Science,2005,282(2):422-427.
[11] 李文华,房晓红,李彬,等.蒙脱石吸附CH4和CO2的分子模拟[J].东北石油大学学报,2014,38(3):25-30.
LI Wenhua,FANG Xiaohong,LI Bin,et al.Molecular simulation of the sorption of methane and carbon dioxide in the montmorillonite[J].Journal of Northeast Petroleum University,2014,38(3):25-30.
[12] 熊健,刘向君,梁利喜.甲烷在蒙脱石狭缝孔中吸附行为的分子模拟[J].石油学报,2016,37(8):1021-1029.
XIONG Jian,LIU Xiangjun,LIANG Lixi.Molecular simulation on the adsorption behaviors of methane in montmorillonite slit pores[J].Acta Petrolei Sinica,2016,37(8):1021-1029.
[13] 隋宏光,姚军.CO2/CH4在干酪根中竞争吸附规律的分子模拟[J].中国石油大学学报(自然科学版),2016,40(2):147-154.
SUI Hongguang,YAO Jun.Molecular simulation of CO2/CH4competitive adsorption in kerogen[J].Journal of China University of Petroleum (Edition of Natural Science),2016,40(2):147-154.
[14] 隋宏光,姚军.页岩黏土矿物CH4/CO2吸附规律的分子模拟[J].东北石油大学学报,2016,40(2):90-98.
SUI Hongguang,YAO Jun.Molecular simulation of CH4/CO2adsorption in clay minerals[J].Journal of Northeast Petroleum University,2016,40(2):90-98.
[15] KADOURA A,NAIR A K N,SUN S.Adsorption of carbon dioxide,methane,and their mixture by montmorillonite in the presence of water[J].Microporous & Mesoporous Materials,2016,225:331-341.
[16] DE S.Molecular computer simulations of the swelling properties and interlayer structure of Cesium montmorillonite[J].Langmuir,1998,14(20):5959-5967.
[17] MARRY V,TURQ P,CARTAILLER T,et al.Microscopic simulation of structure and dynamics of water and counterions in a monohydrated montmorillonite[J].Journal of Chemical Physics,2002,117(7):3454-3463.
[18] 王进,曾凡桂,王军霞.锂-,钠-,钾-水化蒙脱石层间结构的分子动力学模拟[J].化学学报,2006,64(16):1654-1658.
WANG Jin,ZENG Fangui,WANG Junxia.Molecular dynamics simulation studies of interlayered structure in lithium-,sodium- and potassium-montmorillonite hydrate[J].Acta Chimica Sinica,2006,64(16):1654-1658.
[19] 徐加放,付元强,田太行,等.蒙脱石水化机理的分子模拟[J].钻井液与完井液,2012,29(4):1-4.
XU Jiafang,FU Yuanqiang,TIAN Taihang,et al.Molecule simulation on mechanism of montmorillonite hydration[J].Drilling Fluid & Completion Fluid,2012,29(4):1-4.
[20] 秀文. 蒙脱石的性能和应用[J]. 精细化工原料及中间体, 2004(11):10-11.
[21] 文玉华,朱如曾,周富信,等.分子动力学模拟的主要技术[J].力学进展,2003,33(1):65-73.
WEN Yuhua,ZHU Ruceng,ZHOU Fuxin,et al.An overview on molecular dynamics simulation[J].Advances in Mechanics,2003,33(1):65-73.
猜你喜欢
测控技术(2021年10期)2021-12-21 07:10:08
中国民间疗法(2021年5期)2021-06-09 09:21:26
现代塑料加工应用(2021年5期)2021-02-28 08:18:02
上海公路(2018年3期)2018-03-21 05:55:40
光学精密工程(2016年5期)2016-11-07 09:06:01
中国人兽共患病学报(2016年6期)2016-01-30 08:13:10
中国塑料(2015年3期)2015-11-27 03:41:54
环境科技(2015年3期)2015-11-08 12:08:28
西南石油大学学报(自然科学版)(2015年4期)2015-08-20 09:05:20
物理化学学报(2015年5期)2015-02-28 17:35:06
