
毛细管区带电泳测定大黄提取物中有效成分的含量*
2018-02-02 06:45杨元娟刘应杰夏培元
重庆医学 2018年2期
曾 雪,杨元娟,陈 竹,刘应杰,夏培元
(1.重庆医药高等专科学校 401331;2.第三军医大学西南医院药剂科,重庆 400050;3.重庆市药物制剂工程技术研究中心 401331;4.重庆市食品药品检验检测研究院 401121;5.重庆市化学药品质量控制与评价协同创新中心 401121)
药用大黄是我国常用中药,具有多种药理作用[1-3]。有效成分的检测方法主要有纸色谱法、柱色谱法(HPLC)[4]、薄层色谱法[5]和高效液相色谱法[6-8],但液相色谱操作过程复杂,流动相处理要求高,试剂消耗量大。高效毛细管电泳(high performance capillary electrophoresis,HPCE)是一种具有灵敏度和分辨率高、成本较低的分离分析技术[9-10]。本研究以大黄为对象,回流提取有效成分,并采用高效毛细管电泳作为分离和检测手段,通过加入β-环糊精和调节缓冲溶液pH实现更好分离效率,该方法国内尚无相关研究,是液相色谱法的一个有力补充[11-13]。
1 材料与方法
1.1材料 CL1020高效毛细管电泳仪(紫外检测器,北京彩陆仪器有限公司,中科院研究生院应化所,中国),GWA-UN1型超纯水器(北京普析通用仪器有限公司,中国),SK-1涡旋混合器(常州市国旺仪器制造有限公司,中国),AUX220电子天平(岛津,日本)。Na2B4O7·10 H2O(分析纯,重庆北碚精细化工工厂);无水碳酸钠(分析纯,重庆北碚精细化工工厂);羟丙基-β-环糊精(HP-β-CD,相对分子量1 459.8,ACROS ORGANICS,New Jersey,USA);大黄素(C15H10O5,批号must-14110704)、大黄酚(C15H10O4,批号must-14072415)和大黄酸(C15H8O6,批号must-14072401,HPLC纯度大于或等于98.0%)对照品(美仑生物科技有限公司,中国),药用大黄饮片(四川省阿坝州松潘县药用大黄栽培基地),其他试剂均采用分析纯。
1.2方法
1.2.1对照品溶液的制备 精密称取大黄素对照品、大黄酸对照品和大黄酚对照品适量,加甲醇分别制成每1 mL含大黄酸、大黄素、大黄酚各8.0 mg的对照品贮备液;分别精密量取上述对照品贮备液各2 mL,定容至10 mL,混合摇匀,即得(每1 mL中大黄素、大黄酸、大黄酚各1.6 mg)。
1.2.2供试品溶液的制备 取药用大黄饮片粉碎,取粉末(过四号筛)约0.15 g,精密称定,置具塞锥形瓶中,精密加入甲醇25 mL,称定质量,加热回流1 h,放冷,再称定质量,用甲醇补足减失的质量,摇匀,滤过。精密量取续滤液5 mL,置烧瓶中,挥去溶剂,加8%盐酸溶液10 mL,超声处理5 min,再加三氯甲烷10 mL,加热回流1 h,放冷,置分液漏斗中,用少量三氯甲烷洗涤容器,并入分液漏斗中,分取三氯甲烷层,酸液再用三氯甲烷提取3次,每次10 mL,合并三氯甲烷液,减压回收溶剂至干,残渣加甲醇使溶解,转移至10 mL量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得[14-15]。
1.2.3电泳条件
1.2.3.1毛细管预处理 采用67.4 cm×75.0 μm石英毛细管柱(有效长度51.0 cm,河北永年瑞丰色谱配件有限公司)为分离柱,毛细管分别用0.1 mol/L NaOH冲洗活化10 min,水洗5 min,再用缓冲溶液冲洗5 min。每次进样前都分别用0.1 mol/L NaOH冲洗3 min,水洗1 min,再用缓冲溶液冲洗2 min,以保证迁移时间和峰面积的重现性。
1.2.3.2电泳条件设定 分离电压范围:15~30 kV,进样方式采用压差进样,进样时间5 s,操作温度25 ℃,缓冲溶液:60 mmol/L Na2B4O7+40 mmol/L Na2CO3+40 mmol/L HP-β-CD(pH 8.9);供试品缓冲溶液与此相同。
2 结 果
2.1电泳条件优化
2.1.1检测波长的选择 大黄素和大黄酸的吸收波长均在250~270 nm处,其中254 nm为最大吸收波长。大黄酚在254、530 nm处有最大吸收,依据中国药典2015版,均采用254 nm为检测波长,因此本实验采用254 nm波长为检测波长。
2.1.2进样方式的选择 毛细管电泳的进样方式对分离效率和重现性有重要影响,压力进样和电动进样为最常见的两种进样方式,电动进样对粘度较大的生化样品比较适合,但测量重现性较差,且可能因各组分扩散系数的差别而导致迁移速率的改变,本试验的标本为大黄提取物,相对生化样品粘度较低,且注重重现性,因此选择压力进样作为进样方式。采用进样压力0.5 psi,进样实践0.5 s。
2.1.3缓冲溶液及浓度的优化 由于大黄素、大黄酚和大黄酸的结构近似,电荷差异小,且大黄提取中成分较多,采用区带电泳分离模式(CZE)较难实现3种目标组分的完全分离,因此本试验通过在缓冲体系中加入低浓度HP-β-CD,在传统的CZE电泳模式下引入胶束电色谱模式,利用3组分在分配系数上的差异,当HP-β-CD浓度达到40 mmol/L时,实现完全分离,见图1、2。

A:大黄素;B:大黄酚;C:大黄酸
图1大黄主要活性成分结构式
2.2方法学验证
2.2.1精密度试验 精密量取“1.2.1”项下对照品溶液,按“1.2.3.1”项下电泳条件连续进样测定3次,记录峰面积。大黄素、大黄酚和大黄酸相对标准偏差(RSD)分别是 1.79%、4.46%和2.30%,表明仪器精密度良好,见表1。
2.2.2线性范围 分别精密吸取“1.2.1”项下大黄素、大黄酚和大黄酸对照品溶液2 mL于10 mL容量瓶中定容,配置成浓度为1.6 mg/mL对照品溶液,再分别吸取对照溶液0.3、0.8、1.3、1.8、2.3、2.8 mL,置于6个10 mL容量瓶中,用70%甲醇定容至刻度,摇匀,得等浓度梯度对照品溶液,并分别以“1.2.3”项下电泳条件进行电泳分析检测,分别以3组分对照品浓度为横坐标(X),以对照品峰面积为纵坐标(Y)进行回归计算,得到大黄素回归方程为:Y=0.079 4X-0.034 6,r=0.995 7;大黄酚回归方程为:Y=0.085 3X-0.042 8,r=0.995 9;大黄酸回归方程为:Y=0.078 4X-0.073 2,r=0.995 4,鉴于压力进样存在一定误差,3组分共同线性范围在1.1~19.0 mg/mL范围内线性关系良好。
2.2.3回收率试验 精密称取大黄样品粉末约1.5 g,共3份,分别置于5个锥形瓶中,按“1.2.2”项下“大黄提取物供试品”方法制备供试品溶液,先按“1.2.3”项下电泳条件检测并记录每份样品的电泳谱图,再在每份供试品中加入等量的大黄素、大黄酚和大黄酸对照品各3 mg,再次测定其含量,分别计算大黄素、大黄酚和大黄酸的平均回收率,结果显示该方法单组分回收率较好,多组分RSD偏高,分析可能是由于大黄提取物成分较多,且本方法是同时考察3组分在电泳中含量,目标组分间存在化学性质差异,导致检测效率不一致所致,见表2。

A:CZE 模式下CE分离检测;B:HP-β-CD修饰后CZE模式下大黄混合标品CE分离检测;C:HP-β-CD修饰后CZE模式下大黄提取物CE分离检测

图2 HP-β-CD修饰前后大黄毛细管电泳对比图
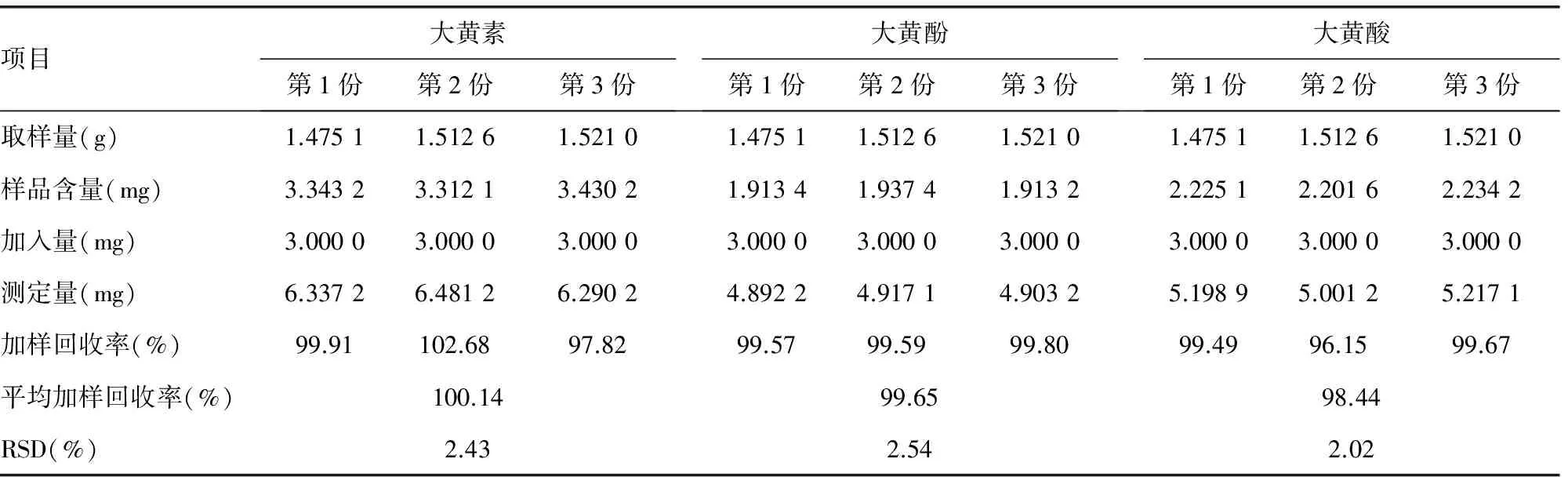
表2 回收率试验结果(n=3)
2.3样品含量测定 对3批样品进行含量测定,分别精密称取不同批次大黄药材样品粉末约1.5 g,按“1.2.2”项下“大黄提取物供试品”方法制备供试品溶液,再按“1.2.3”项下电泳条件进行电泳分离检测,分别记录大黄素、大黄酚和大黄酸含量,见表3。
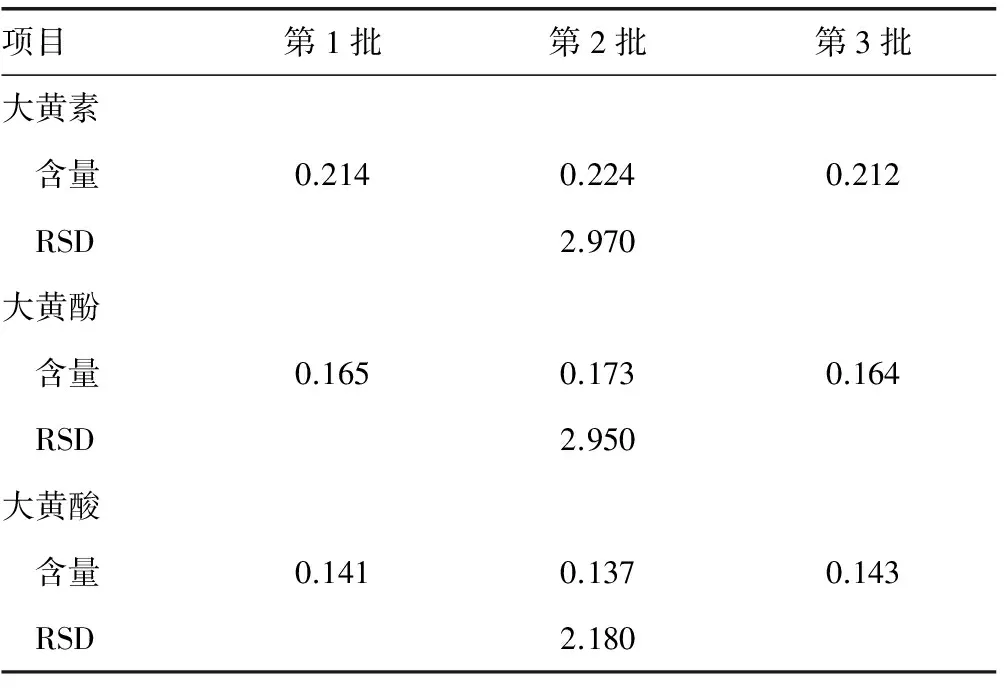
表3 3批次样品中大黄提取物的含量测定结果(n=3,%)
3 讨 论
大黄素、大黄酚、大黄酸3组分化学结构相似,带电差异小,CZE模式分离效果较差,本文加入HP-β-CD是因为环糊精分子略呈锥形的中空圆筒立体环状结构,具有一定的疏水性,增加了普通缓冲溶液的多相特性,待测组分在环糊精修饰的缓冲体系中,由于不同的疏水性而呈现出不同的分配系数,从而使不同组分在毛细管电泳中不仅受到电渗流的影响,同时也受到分配色谱的影响,有利于组分的完全分离。
电泳进样方式对本方法的重现性和稳定性有很大影响,电动进样操作简便且进样量稳定,但更适合于粘度较大的生化样品,而本文是中药提取物且带电性不明显,用电动进样不利于样品的导入,所以采用压力进样,保证了进样的重现性。
方法学考察中含量测定的RSD偏高,分析原因是因为本方法优先考虑大黄提取物多组分的分离度,在保证完全分离基础上,牺牲了部分检测准确度,导致结果RSD偏高,可在后期方法优化上进一步改进。
通过β-环糊精修饰毛细管区带电泳测定大黄提取物的方法分析速度快,缓冲溶液简单,试剂消耗量少,可作为高效液相法的重要补充。
[1]刘晗,高云.大黄素药理作用的分子机制研究进展[J].中国药理学通报,2009,25(12):1552-1555.
[2]SUNEELA D,DIPMALA P.Synthesis and pharmacokinetic profile of rhein- boswellic acid conjugate[J].Bioorg Med Chem Lett,2012,22(24):7582-7587.
[3]纪春梅,甄永占,骆广玲,等.赖氨大黄酸通过升高miRNA-451抑制肺癌A549细胞迁移[J].第三军医大学学报,2014,36(3):248-252.
[4]赵东梅,王威,刘坤,等.柱色谱-高速逆流色谱法分离纯化虎杖中大黄素-8-O-β-D-吡喃葡萄糖苷[J].中草药,2016,47(12):2118-2122.
[5]王晓剑,张悠,王晓红.大黄散质量标准研究[J].山西医科大学学报,2016,47(11):988-991.
[6]马惠玲,谢鑫,麦曦,等.高效液相色谱法测定黄连上清丸中芦荟大黄素和大黄酸的含量[J].南昌大学学报(医学版),2016,56(3):16-19.
[7]潘丽丽,李亭亭,杨颂,等.HPLC法同时测定牛黄解毒片中黄芩苷、黄芩素、芦荟大黄素、大黄素的含量[J].中医药学报,2015,43(1):50-53.
[8]李莉,宋俊骊,王志梅,等.高效液相色谱法同时测定九制大黄丸中5组分含量[J].中国药业,2015,24(5):34-36.
[9]刘会前.HPLC法测定保肾灵I号中大黄素和大黄酚的含量[J].重庆医学,2005,34(11):1703-1704.
[10]郭芳芳,冯锋,白云峰,等.高效毛细管电泳用于检测黄花菜中4种活性物质[J].食品科学,2016,37(4):73-76.
[11]陈琴华,李鹏,朱军.毛细管电泳技术在黄酮类化合物分析中的应用进展[J].医药导报,2012,31(10):1329-1333.
[12]于虹敏,林文津,徐榕青,等.毛细管电泳技术在药学研究的应用概述[J].中国民族民间医药,2012,21(1):34.
[13]王德先,赵敬湘,杨更亮,等.毛细管区带电泳法测定中药金银花中绿原酸的含量[J].中草药,2000,31(6):432-434.
[14]杨世颖,刘淑聪,杜冠华,等.大黄中药材及其醇、水提取物中大黄素成分分析标准物质研究[J].中国中药杂志,2016,41(3):456-462.
[15]林海丹,翟海云,周清.毛细管电泳法同时测定牛黄解毒片中大黄酸和大黄素的含量[J].中药材,2012,35(6):992-993.
猜你喜欢
世界最新医学信息文摘(2021年12期)2021-06-09
云南化工(2020年11期)2021-01-14
化学与粘合(2020年6期)2020-03-08
装备制造技术(2019年12期)2019-12-25
科技视界(2017年25期)2017-12-11
现代检验医学杂志(2016年1期)2016-11-12
广州化工(2016年1期)2016-09-01
杂草学报(2015年2期)2016-01-04
中药与临床(2015年5期)2015-12-17
食品工业科技(2014年5期)2014-03-11
