
沉积物中重金属形态分析技术的适用范围
2018-01-22 08:03毛凌晨李飞鹏
理化检验-化学分册 2017年9期
毛凌晨,施 柳,叶 华,林 童,李飞鹏,陶 红
(上海理工大学 环境与建筑学院,上海200093)
沉积物是水环境中重金属的源和汇。经过工农业废水排放、大气沉降和地表径流等途径进入水体的重金属,会优先吸附到颗粒物上并伴随颗粒物进入沉积物[1-2]。在一定的外界条件变化下,如酸度、氧化还原条件、盐度等[3],这些金属元素又会通过不断的吸附与解吸过程形成二次污染[4]。研究表明:沉积物中重金属的环境行为与生物毒性主要取决于其化学与物理形态[5-6],同时环境条件变化也会促使重金属形态发生转化[7]。因此,研究重金属的赋存形态对于沉积物的环境风险评估与治理修复方法的选择尤其重要[6]。
1 单一提取法
单一提取法是采用特定的某种或多种混合提取剂,通过一步提取的过程,释放出特定沉积物相或结合态的元素[15]。该方法简便、可操作性强,便于快速判断沉积物的污染程度,预测重金属离子的活性,从而分析重金属形态对环境的潜在危害性。提取剂一般可分为水、稀酸溶液、盐溶液、螯合剂与混合溶液等五类,各有其适用范围。另外,提取结果也会随着提取剂的化学性质与浓度、固液比、振荡速率、温度和提取时间的不同而变化[16]。因此,在操作前,应对试验目的、目标元素、样品理化性质、检测手段与分析仪器等有全面的认识,选用适当的提取剂与操作方法。
使用水作为提取剂可提取的水溶态元素是沉积物中活性最强的组分,包括水溶性离子、可溶态有机质及其他组分结合的离子[14]。当前,随着高精密分析仪器的发展,以去离子水作提取剂越来越多被使用于测定元素在环境中的迁移性和生物毒性。但是,这种方法在检测分析上的局限性仍较明显,原因在于:① 由于水本身不具备酸碱缓冲能力,因此提取过程中系统的酸度无法得到有效控制;② 被溶解的金属离子会产生较严重的再吸附现象,直接影响提取结果[17];③ 离子强度很低,致使重金属容易吸附在过滤膜表面或者发生胶体浸透现象;④ 一般需要72h的平衡时间,试验过程较长[18-19];⑤ 水提取态元素浓度通常很低,应选用灵敏度高、检出限低的电感耦合等离子体质谱法(ICP-MS)等进行测定,不适用于元素溶解度很低的碱性沉积物。在试验设计时,应发挥水作为提取剂不会对自然环境产生影响的优势,将其应用于沉积物的原位监测,并配以过滤、渗析等尺寸分离方法,可得重金属在水溶态的形态分布。
稀酸溶液提取沉积物中重金属元素时,典型的提取剂有硝酸、盐酸和乙酸。稀酸能溶解一系列不同形态的重金属元素,如可交换态、碳酸盐结合态、无定型铁锰氧化物结合态和有机物结合态等。中国地质调查局颁布的《2005-3生态地球化学评价样品分析技术要求》规定使用稀盐酸(或稀硝酸)作为酸、中性土壤中铅的可浸提量的提取剂。其中,选用硝酸作为提取剂在提取金属的过程中不会对平衡离子产生干扰[14],在检测上具有一定的优势。研究表明,稀硝酸可有效提取酸性与中性的土壤中活性重金属,并用以计算重金属元素在土壤溶液中的溶解度与形态分布[20-22];对于pH 较高的石灰质土壤,当重金属总量较高,并且其中只有少量为活性金属时,硝酸往往会提取部分稳定态的重金属[18,23]。
使用中性盐(如CaCl2、NaNO3、MgCl2等)溶液提取是获取沉积物中活性金属元素的常用方法。沉积物重金属的提取剂应优先考虑0.01mol·L-1CaCl2溶液,因其具有以下优势[24-25]:① 不会影响交换位的酸度,也不会作用于硅酸盐或氢氧化物,对沉积物酸碱性影响小;② 二价阳离子可以絮凝沉淀悬浊液中的胶体,降低其对检测结果的干扰;③0.01mol·L-1Ca2+强度与典型沉积物相当。这些特点保证了0.01mol·L-1CaCl2溶液能有效置换出被沉积物颗粒吸附的活性金属离子,常用于提取由于静电引力作用而存在于沉积物表面带电荷的可交换性微/痕量元素。若提取液中重金属浓度过低,可以考虑选用1mol·L-1乙酸铵溶液[26]。但是,如果沉积物中碳酸盐含量较高,则需加入缓冲剂保持pH为7左右,防止碳酸盐被溶解[27-28]。值得注意的是,在使用物理光谱法测定金属时,提取剂中的阳离子容易引起干扰而影响测定结果[14],因此应在保证目标元素浓度高于检出限的情况下,尽可能稀释进样溶液,减少干扰。
乙二胺四乙酸(EDTA)和二乙烯三胺五乙酸(DTPA)等螯合剂能与沉积物释放的重金属形成螯合物而稳定地存在提取液中,提取力度要强于中性盐。多数研究者认为,浓度恰当的EDTA溶液可能是单一提取法中预估多种金属活性的最佳提取剂[29-32]。DEGRYSE 等[32]建议 EDTA 的用量应为50~100mmol·kg-1(土壤或沉积物)。EDTA用量过低可能导致提取不完全,用量过高则会释放非活性的金属[33]。在碱性环境的沉积物样品中,EDTA可能会释放部分非活性的金属,如Ni、Cu、Cd和 Pb[31,33-34]等。另外,由于 EDTA 无法与一些 元素(如 Mo和 Ti)形成稳定的化合物[31],与 Cr3+的络合反应很慢(大于5d)[31],不适用于这3种元素的分析。DTPA(常用浓度0.005mol·L-1)作为较弱的提取剂,由于较难形成化合物,在碱性较高的沉积物中,可以避免过量提取[35];然而,在有些情况下,也会因此低估了其活性[34]。综上所述,采用螯合剂提取时,应该在试验前首先确认目标元素与EDTA、DTPA的络合强度,以保证结果的准确性。
混合通用浸提剂主要用于土壤有效态元素的提取分析,比较常用的有 Mehlich 1试剂(简称 M1)、Mehlich 3试剂(简称 M3)、Morgan-Wolf试剂和Soltanpour-Swab试剂等。研究发现,M3(HOAc-NH4NO3-NH4F-HNO3-EDTA,pH 2.5)能适用于各类酸性、中性和碱性土壤[36]。当前多数研究将M3用于有效提取P、Mg、Ca等主要营养元素中。马立锋等[37]和JOSHI等[38]的试验结果均表明,M3对Zn、Pb、Cd、Cr、Ni和Co等重金属元素的提取结果普遍大于DTPA和0.1mol·L-1盐酸溶液的提取结果。WANG等[39]比较了分别使用DTPA、M3和乙酸提取沉积物中重金属的有效性,特别是生菜、萝卜与大豆中的Cd、Cr、Hg和Pb,发现M3更适用于预测根茎类植物中的重金属总量。
总体来说,以上提取方法各有优劣势,试剂的选用、试剂用量和提取时间是主要的影响因素。碱性沉积物在检测上难度较高,应慎用弱酸,此时可选用浓度较低的EDTA溶液,以保证提取液中的金属浓度能满足测定要求。单一提取法的适用范围以及注意事项见表1。
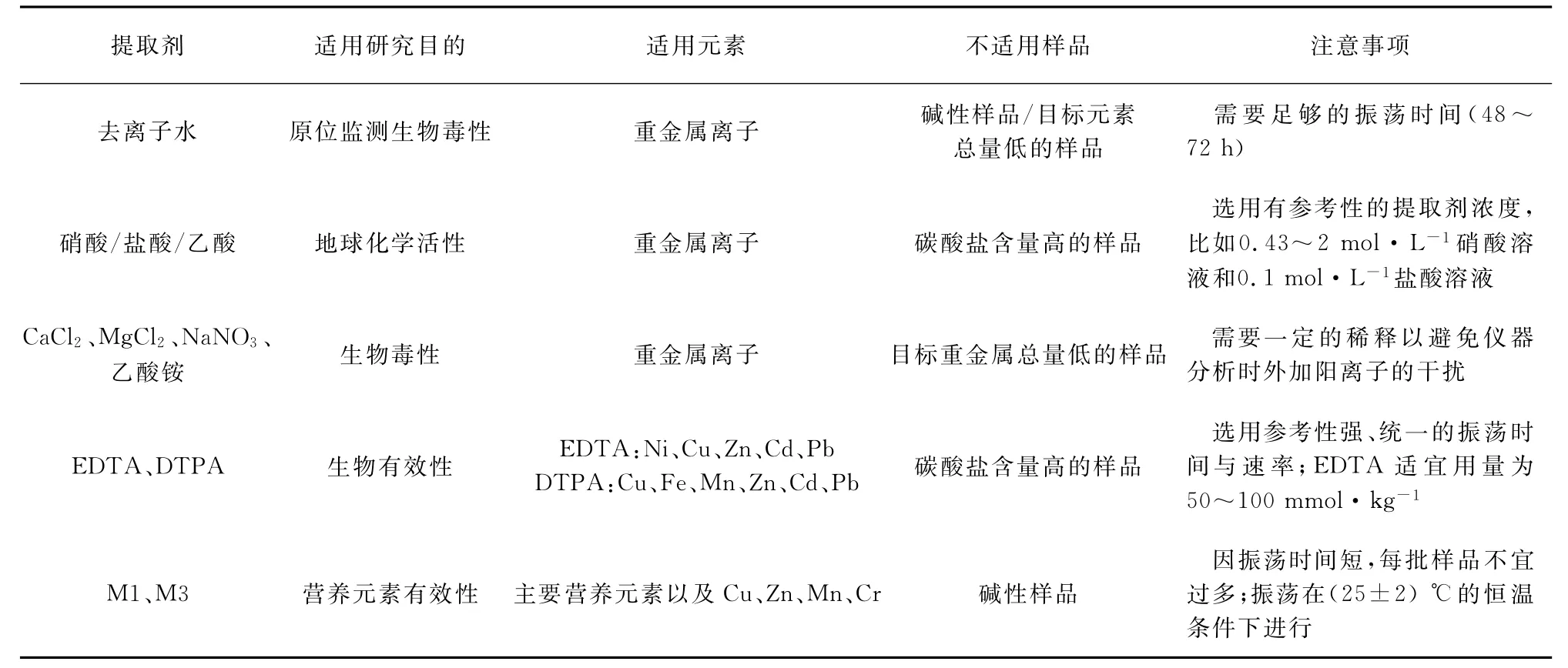
表1 单一提取法的应用Tab.1 Application of single extraction method
2 连续提取法
典型的连续提取法是使用一系列提取力度渐增的提取剂,提取沉积物中不同化学形态的金属,并应用ICP-MS等仪器测定各形态的含量。表2按提取步数列举了几种经典的连续提取法,其中目前应用最广泛的为TESSIER[40]提出的五步法和欧共体物质标准局(European Communities Bureau of Reference)提出的 BCR 四步法[41],涉及潮滩[42-44]、河道底泥[45-47]、湿地与湖泊等环境的沉积物[48-49]。

表2 几种经典的沉积物中重金属连续提取法Tab.2 Several classcial sequential extraction procedures for heavy metals in sediments
较单一提取法而言,连续提取法的优势在于能够提供更多的元素形态信息,做出更准确的生态风险评估。以TESSIER法为例,其对于沉积物重金属的提取结果可以同时用于预估生物毒性(第一步),生物有效性(第一、二步)以及地球化学活性(第一至四步),通过这三部分之间的关系,可进而建立模型以探讨元素形态在复杂环境中的转化机制。此外,结合其他分析手段,比如同位素示踪技术,连续提取法可被应用于探究重金属在纵向或平面上的迁移转化[50-52];或者在对受污染沉积物进行修复时,借助X-射线衍射(XRD)、扫描电子显微镜(SEM)等定性分析手段,对照修复前后污染物的形态变化,研究修复机理与效果[53-55]。同时,连续提取法可以根据研究的需要灵活调整,例如,当样品中氧化物含量较高时,则可通过加入不同还原强度的试剂,将氧化锰、非晶型或晶型氧化物分离[56-58]。不同形态的铁锰氧化物对元素的结合强度因其结构不同而存在明显差异。对重金属元素而言,主要表现在:① 非晶型氧化物的表面活性较强;② 晶型氧化物中的重金属则很难被释放出来;③ 氧化锰对金属离子的结合力要大于铁氧化物。
然而,在实际操作中,连续提取法的局限性日趋明显。由于提取剂的选择性弱,无法确保在每一步中能够准确提取对应形态的金属,而导致试验误差[14],主要体现在三个方面。第一,每一种试剂都会提取其他形态的元素,特别是还原剂对有机结合态的影响[59]。因此有研究指出,对有机物结合态的提取应该在铁锰氧化物之前[14]。有些较强的提取剂,比如DCB(柠檬酸钠、碳酸氢钠和连二亚硫酸钠),甚至能提取硅酸盐晶体中(即残渣态)的部分重金属元素[40]。第二,对某一形态的元素提取不完全。SCHRAMEL等[60]发现在BCR法的第二步中,盐酸羟胺对于晶型氧化铁的提取并不完全。第三,由于前一步中加入的提取剂改变了整个系统的酸度,被提取的金属会发生再吸附而导致检测误差[61]。针对这些问题,一些研究者对方法进行了改进,以减少每一步之间的相互干扰,提高对各个形态的选择性[62]。除了方法本身的局限性,在与其他方法所得结果进行比较时,越来越多的研究者发现连续提取法的作用在风险评估与生物有效性研究中可能十分有限。例如,与同位素稀释技术所测得土壤中活性Cd与Pb含量的比较,发现重金属活性与连续提取法所得的任何一个形态都没有相关性[63-65];与植物试验比较发现,根部所能吸收的元素含量仅限于第一步所提取的形态[66]。
在沉积物重金属研究中,连续提取法若使用得当,其结果仍然具有很高的参考价值[66]。考虑连续提取法的优势和局限性,在实际研究中可从以下几个方面加以考虑:① 应用于“比较性”研究,比如时间与空间上重金属形态的差异、修复处理前后的形态变化等。或者应考虑加入其他非化学提取类方法作为形态分析结果的参考比对,如同位素稀释、植物试验等。② 选择“标准”方法。由于研究者竞相将经典方法进行改良,使得这些不同方法所得的试验结果无法有效共享。因此,虽然目前研究者对于连续提取法标准化的问题依旧存在争议,但可以优先考虑经典的TESSIER法和BCR法,特别是有标准物质做质量控制的方法[14],加强与其他研究结果进行更准确的比较研究。③ 做结论时应谨慎。鉴于连续提取法不可能准确提取某种形态的重金属,在结论中应选择使用“操作性定义”的词,如有机结合态为可氧化态,在研究重金属活性与生物有效性时也需措辞严谨[14]。④ 经济与时间成本。由于连续提取法对精度要求很高,必须使用超纯试剂,而且有些步骤中反应时间长。虽然步骤越多,所得的信息越丰富,但也必然增加人力物力成本和误差几率,因此应明确目的,选择有针对性的方法。⑤ 恰当的保存和预处理。环境条件的变化会对元素的形态产生显著影响,特别是沉积物样品[10],当含水率改变时,氧化-还原条件改变,造成铁锰氧化物的溶解或结晶,会直接影响铁锰氧化物结合态与溶解态金属的含量[67-68]。⑥ 在操作中,应尽可能减少前一步提取遗留在样品里的提取液。
3 基于粒度的形态分类法
受水动力扰动的影响,沉积物颗粒、胶体粒度对重金属在水体中的迁移与空间分布影响显著[13]。因此,基于粒度的重金属形态分类方法在环境研究中也倍受重视。常见的分类方法有按土壤颗粒粒级(即黏粒、粉粒和砂粒)过筛[70-72]或膜过滤[73-74]。过筛后的颗粒需进行化学提取或消解,随后使用光/质谱法测定每个粒级沉积物中相关元素含量。膜过滤常用的粒径阈值有0.2μm和0.45μm,过滤后的渗滤液中(或保留在过滤器上)的重金属含量一般可以直接使用光/质谱法测定。
由于沉积物颗粒表面的吸附作用,重金属含量通常会随着粒度减小(比表面积增大)而增大[75]。因此,对于细小颗粒中重金属分布的研究尤为重要。王永红等[13]对细颗粒物(直径<63μm)中重金属的研究做了总结。近年来,国内外环境化学研究逐渐开始关注胶体结合态(<0.2μm)中重金属的迁移性及其对沉积物-水体的影响[76-77]。针对粒径在0.2μm以下的胶体和大分子的分离方法相继被开发。使用最 为 广 泛 的 有超 滤[78-79]、场 流 分 级 法(FFF)[76,80-81]和尺寸排阻色谱法(SEC)[76,82],这些技术的优势在于,样品被分级分离后可以直接连接到ICP光/质谱仪上进行检测,减少了在实验室中被污染的几率。其中超滤法与普通过滤法在分离粒级上明显不同,但分离原理上并无本质区别。而FFF与SEC属于(类)色谱法,能提供连续的粒度分布结果,相较而言FFF所能分析的粒径范围(1nm~1μm)大于SEC,分离度也更高[14,83]。
尽管这三种技术所能分离的粒径都达到了纳米级别,但与化学提取法相同,也只能根据离子结合强弱与分布提供在操作意义上的重金属分布与形态信息,无法给出真正意义上的重金属分子化学形态。同时分级分离方法和材料的选择也会极大地影响研究结果。COLLINS[84]用色谱法成功测定了与有机酸小分子上的二、三羧酸结合的Fe、Ni和Co;然而,JACKSON 等[76]和 MAO 等[82]在使用 SEC 分离有机物大分子时,分别发现Ni和Cu均会受到柱体移动相液体(即浸提液)不同程度的影响,在分离过程中从有机物大分子上脱离,造成分析误差。HASSELLOV 等[85]和 DUBASCOUX 等[86]认为在使用FFF分离的过程中,目标金属与胶体之间的连接必须避免其他介质的影响,才能成功使用这种技术对胶体态重金属进行定量分析。因此,应该谨慎选择作为分离介质(即FFF技术中的膜与SEC技术中的填柱凝胶)与提取试剂,确保目标金属不会与它们发生反应而影响试验结果。
虽然FFF与SEC的分离原理不同,但在浸提液的选择上原理相同,即① 不会促使胶体产生絮凝或分解现象;② 不改变胶体的流体直径;③ 不与胶体态的目标金属发生反应。基于前两点,在环境研究中宜多选用单价盐作为提取液。针对第三点,作为保障定量分析胶体态重金属的关键,则因金属本身属性而异。
表3列举了近年来使用SEC测定胶体态重金属的研究报道,从表中可以发现,使用较广泛的提取剂有NH4NO3、NH4Cl和Tris缓冲剂。
4 结论
综上所述,被广泛应用于各种沉积物环境研究中化学形态提取法与基于粒度的形态分离方法均属于“操作性定义”,虽然实验室操作性强,但都不能真正量化重金属的各形态。因此,建议将化学提取法应用于沉积物重金属的迁移转化等“比较性”研究。在分析重金属的生物毒性、生物有效性等因素时应选用参考性强的方法,即应用较广泛的提取剂浓度、连续提取的步骤等。试验分析前应通过预试验了解样品的主要理化性质,特别是酸度、氧化还原电位等,以便在样品保存、预处理以及试验过程中尽可能减少重金属形态发生改变的机率与程度。
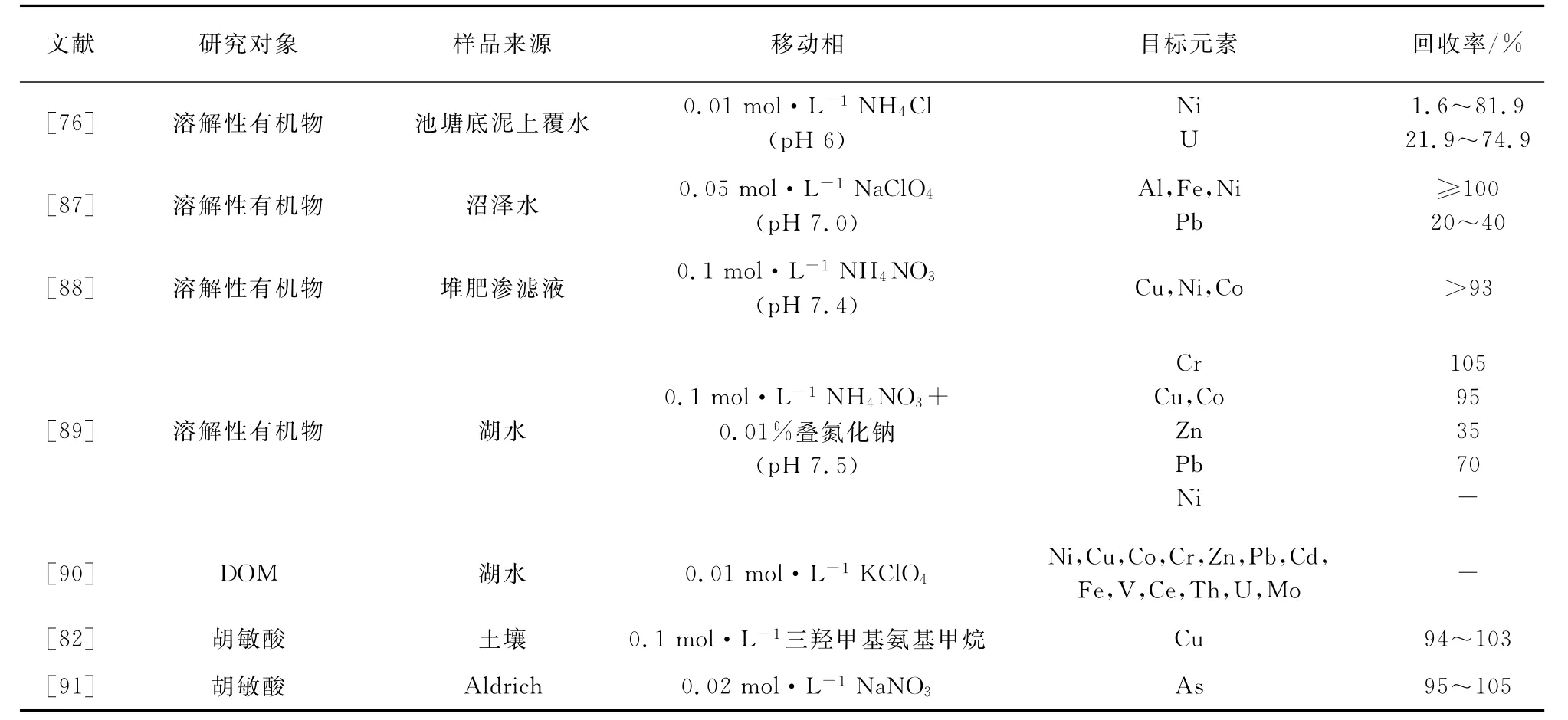
表3 尺寸排阻色谱法测定胶体重金属研究报道Tab.3 Researchs on exclusion chromatography technique used for colloid-metal studies
[1] LAFABRIE C,PERGENT G,KANTIN R,et al.Chemosphere[J],2007,68(11):2033-2039.
[2] 陈春霄,姜霞,战玉柱,等.中国环境科学[J],2011(11):1842-1848.
[3] ZHU H N,YUAN X Z,ZENG G M,et al.Transactions of Nonferrous Metals Society of China[J],2012,22(6):1470-1477.
[4] 孟伟,刘征涛,范薇.环境科学研究[J],2004(6):66-69.
[5] CANCES B,PONTHIEU M,CASTREC-ROUELLE M,et al.Geoderma[J],2003,113(3/4):341-355.
[6] YANG Z,WANG Y,SHEN Z,et al.Journal of Hazardous Materials[J],2009,166(2/3):1186-1194.
[7] LI H,QIAN X,HU W,et al.Science of the Total Environment[J],2013,456:212-221.
[8] 冯素萍,鞠莉,沈永,等.化学分析计量[J],2006(4):72-74.
[9] 李非里,刘丛强,宋照亮.中国环境监测[J],2005(4):21-27.
[10] 关天霞,何红波,张旭东,等.土壤通报[J],2011(2):503-512.
[11] 陈怀满.环境土壤学[M].北京:科学出版社,2010.
[12] 刘素美,张经.东海海洋[J],1998(3):49-56.
[13] 王永红,张经,沈焕庭.地球科学进展[J],2002(1):69-77.
[14] LAING G D.Trace Elements in Soils[M].Chichester:Wiley,2010.
[15] 周国华.物探与化探[J],2014(6):1097-1106.
[16] BECKETT P H T.Advances in Soil Science[M].New York,London:Springer-Verlag,1989.
[17] GLEYZES C,TELLIER S,ASTRUC M.Trac-Trends in Analytical Chemistry[J],2002,21(6/7):451-467.
[18] SINAJ S,MACHLER F,FROSSARD E.Soil Science Society of America Journal[J],1999,63(6):1618-1625.
[19] MEERS E,UNAMUNO V R,LAING G D,et al.Geoderma[J],2006,136(1/2):107-119.
[20] ALMAS A R,LOFTS S,MULDER J,et al.European Journal of Soil Science[J],2007,58(5):1074-1086.
[21] TIPPING E,RIEUWERTS J,PAN G,et al.Environmental Pollution[J],2003,125(2):213-225.
[22] WENG L P,TEMMINGHOFF E J M,VAN RIEMSDIJK W H.Environmental Science and Technology[J],2001,35(22):4436-4443.
[23] QIAN J,SHAN X Q,WANG Z J,et al.Science of the Total Environment[J],1996,187(2):131-141.
[24] HOUBA V J G,LEXMOND T M,NOVOZAMSKY I,et al.Science of the Total Environment[J],1996,178(1/3):21-28.
[25] HOUBA V J G,TEMMINGHOFF E J M,GAIK-HORST G A,et al.Communications in Soil Science and Plant Analysis[J],2000,31(9/10):1299-1396.
[26] HLAVAY J,POLY K K.Handbook of elemental speciation:Techniques and methodology[M].Chichester:Wiley,2003.
[27] BAKER A J M,REEVES R D,HAJAR A S M.New Phytol[J],1994,127:61-68.
[28] GOMMY C,PERDRIX E,GALLOO J C,et al.International Journal of Environmental Analytical Chemistry[J],1998,72(1):27-45.
[29] WELP G,BRUMMER G W.Journal of Plant Nutrition and Soil Science-Zeitschrift Fur Pflanzenernahrung Und Bodenkunde[J],1999,162(2):155-161.
[30] TONGTAVEE N,SHIOWATANA J,MCLAREN R G,et al.Science of the Total Environment[J],2005,348(1/3):244-256.
[31] GABLER H E,BAHR A,HEIDKAMP A,et al.European Journal of Soil Science[J],2007,58(3):746-757.
[32] DEGRYSE F,SMOLDERS E,PARKER D R.European Journal of Soil Science[J],2009,60(4):590-612.
[33] DEGRYSE F,BUEKERS J,SMOLDERS E.European Journal of Soil Science[J],2004,55(1):113-121.
[34] MARZOUK E R,CHENERY S R,YOUNG S D.European Journal of Soil Science[J],2013,64(4):526-536.
[35] LINDSAY W L,NORVELL W A.Soil Science Society of America Journal[J],1978,42(3):421-428.
[36] 于群英,段立珍.安徽农业科学[J],2002(6):861-862.
[37] 马立锋,杨亦扬,石元值,等.土壤通报[J],2007(4):745-748.
[38] JOSHI D,SRIVASTAVA P C,DWIVEDI R,et al.Chemical Speciation and Bioavailability[J],2014,26(3):148-157.
[39] WANG T,SUN H,MAO H,et al.Journal of Hazardous Materials[J],2014,278:483-490.
[40] TESSIER A,CAMPBELL P G C,BISSON M.Analytical Chemistry[J],1979,51(7):844-851.
[41] URE A M,QUEVAUVILLER P,MUNTAU H,et al.International Journal of Environmental Analytical Chemistry[J],1993,51(1/4):135-151.
[42] FAN W,XU Z,WANG W X.Environmental Pollution[J],2014,191:50-57.
[43] YUAN X,ZHANG L,LI J,et al.Catena[J],2014,119:52-60.
[44] LIU H,LIU G,DA C,et al.Journal of Environmental Quality[J],2015,44(1):174-182.
[45] LIN C,HE M,LIU X,et al.Environmental Earth Sciences[J],2013,70(7):3163-3173.
[46] 李如忠,徐晶晶,姜艳敏,等.环境科学研究[J],2013(1):88-96.
[47] WANG J,LIU G,LU L,et al.Environmental Monitoring and Assessment[J],2016,188(1):3-3.
[48] 王书航,王雯雯,姜霞,等.环境科学[J],2013(9):3562-3571.
[49] YE H,ZANG S,XIAO H,et al.International Journal of Environmental Science and Technology[J],2015,12(1):115-124.
[50] TEUTSCH N, EREL Y, HALICZ L,et al.Geochimica et Cosmochimica Acta[J],2001,65(17):2853-2864.
[51] EMMANUEL S,EREL Y.Geochimica et Cosmochimica Acta[J],2002,66(14):2517-2527.
[52] SHIKAZONO N,TATEWAKI K,MOHIUDDIN K M,et al.Environmental Geochemistry and Health[J],2012,34:13-26.
[53] TOMASEVIC D D,DALMACIJA M B,PRICA M D,et al.Chemosphere[J],2013,92(11):1490-1497.
[54] DONI S,MACCI C,PERUZZI E,et al.Ecological Engineering[J],2015,81:146-157.
[55] DERMONT G, BERGERON M, RICHERLAFLECHE M,et al.Science of the Total Environment[J],2010,408(5):1199-1211.
[56] MCLAREN R G,CRAWFORD D V.Journal of Soil Science[J],1973,24(2):172-181.
[57] MILLER W P,MARTENS D C,ZELAZNY L W.Soil Science Society of America Journal[J],1986,50(3):598-601.
[58] KRISHNAMURTI G S R,HUANG P M,VANREES K C J,et al.Analyst[J],1995,120(3):659-665.
[59] AHNSTROM Z S,PARKER D R.Soil Science Society of America Journal[J],1999,63(6):1650-1658.
[60] SCHRAMEL O,MICHALKE B,KETTRUP A.Science of the Total Environment[J],2000,263(1/3):11-22.
[61] CLEVENGER T E.Water,Air and Soil Pollution[J],1990,50(3/4):241-254.
[62] LI X D,THORNTON I.Applied Geochemistry[J],2001,16(15):1693-1706.
[63] AHNSTROM Z A S,PARKER D R.Environmental Science and Technology[J],2001,35(1):121-126.
[64] ATKINSON N R,BAILEY E H,TYE A M,et al.Environmental Chemistry[J],2011,8(5):493-500.
[65] MAO L C,BAILEY E H,CHESTER J,et al.Environmental Chemistry[J],2014,11(6):690-701.
[66] HOODA P S.Trace Elements in Soils[M].Oxford:Wiley-Blackwell,2010.
[67] ZHANG S Z,WANG S X,SHAN X Q.Chemical Speciation and Bioavailability[J],2001,13(3):69-74.
[68] LAING G,RINKLEBE J,VANDECASTEELE B,et al.Science of the Total Environment[J],2009,407(13):3972-3985.
[69] ELLIOTT H A,DEMPSEY B A,MAILLE P J.Journal of Environmental Quality[J],1990,19(2):330-334.
[70] HILLIER S,SUZUKI K,COTTER-HOWELLS J.Applied Geochemistry[J],2001,16(6):597-608.
[71] LIN J G,CHEN S Y,SU C R.Water Science and Technology[J],2003,47(7/8):233-241.
[72] PACIFICO R,ADAMO P,CREMISINI C,et al.Journal of Soils and Sediments[J],2007,7(5):313-325.
[73] MAGI E,IANNI C,SOGGIA F,et al.Journal of Environmental Monitoring[J],2005,7(12):1287-1294.
[74] BUFFLE J,LEPPARD G G.Environmental Science and Technology[J],1995,29(9):2176-2184.
[75] HOROWITZ A J.A Primer on Sediment-Trace Element Chemistry[M].2nd ed.Michigan:Lewis Publishers,1991.
[76] JACKSON B P,RANVILLE J F,BERTSCH P M,et al.Environmental Science and Technology[J],2005,39(8):2478-2485.
[77] GRAHAM N D,STOLL S,LOIZEAU J L.Fundamental and Applied Limnology[J],2014,184(2):87-100.
[78] WANG W S,WEN B,ZHANG S Z,et al.International Journal of Environmental Analytical Chemistry[J],2003,83(5):357-365.
[79] POKROVSKY O S,SHIROKOVA L S,ZABELINA S A,et al.Aquatic Geochemistry[J],2012,18(2):115-139.
[80] SIRIPINYANOND A,BARNES R M,AMARASIRIWARDENA D.Journal of Analytical Atomic Spectrometry[J],2002,17(9):1055-1064.
[81] PLATHE K L,VON DER KAMMER F,HASSELLOV M,et al.Environmental Chemistry[J],2010,7(1):82-93.
[82] MAO L,YOUNG S D,BAILEY E H.Chemosphere[J],2015,131:201-208.
[83] KOWALKOWSKI T,BUSZEWSKI B,CANTADO C,et al.Critical Reviews in Analytical Chemistry[J],2006,36(2):129-135.
[84] COLLINS R N.Journal of Chromatography A[J],2004,1059(1/2):1-12.
[85] HASSELLOV M,KAMMER F V D,BECKETT R.Environmental Colloids and Particles:Behaviour,Separation and Characterisation[M].Chichester:Wiley,2007.
[86] DUBASCOUX S,LE HECHO I,HASSELLOEV M,et al.Journal of Analytical Atomic Spectrometry[J],2010,25(5):613-623.
[87] SCHMITT D,MULLER M B,FRIMMEL F H.Acta Hydrochimica Et Hydrobiologica[J],2001,28(7):400-410.
[88] LABORDA F,RUIZ-BEGUERIA S,BOLEA E,et al.Spectrochimica Acta Part B-Atomic Spectroscopy[J],2009,64(5):392-398.
[89] BOLEA E,GORRIZ M P,BOUBY M,et al.Journal of Chromatography A[J],2006,1129(2):236-246.
[90] WU F C,EVANS D,DILLON P,et al.Journal of Analytical Atomic Spectrometry[J],2004,19(8):979-983.
[91] LIU G L,CAI Y.Chemosphere[J],2010,81(7):890-896.
猜你喜欢
海洋通报(2022年2期)2022-06-30
石油沥青(2022年2期)2022-05-23
海洋石油(2021年3期)2021-11-05
河北环境工程学院学报(2021年1期)2021-03-19
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
农业环境科学学报(2019年12期)2019-12-23
化工环保(2017年3期)2017-06-22
新疆地质(2015年1期)2015-11-18
中国卫生标准管理(2015年14期)2015-01-27
中国果业信息(2015年1期)2015-01-24
