
团簇Mn3BP结构与性质研究
2018-01-18 03:24方志刚崔远东徐诗浩李雯博
辽宁科技大学学报 2017年5期
周 帅,方志刚,崔远东,徐诗浩,刘 琪,冯 天,李雯博
(辽宁科技大学 化学工程学院,辽宁 鞍山 114051)
非晶态合金是近几十年出现的新型金属功能材料,和普通晶态金属合金相比,非晶态合金具有良好的磁学性能[1-3]、较高的耐磨及抗腐蚀性[4-5]、优异的机械强度[6-7]及催化活性[8-12],所以非晶态合金有着很大的研究价值,具有广泛的应用范围,对该类材料的研究是非常有必要的,而对该类材料的研究也是当下的研究热点。对Mn-B二元体系结构、含Mn、P多元体系结构及含Mn、B、P多元体系结构的研究已取得一定进展[13-15],但对Mn-B-P三元体系结构的研究鲜有报道。因此本文将运用密度泛函理论[16-18]研究原子簇Mn3BP的稳定结构及成键性质,旨在对Mn-B-P三元体系结构的性质有更深入的了解和探究其潜在价值。
1 计算方法
原子簇Mn3BP原子序数总和为奇数,所以为二、四重态。根据拓扑学原理[19]设计出原子簇Mn3BP二、四重态的所有可能构型,并采用含相关校正的密度泛函理论(DFT)方法,在B3LYP/Lan12dz水平下对原子簇Mn3BP的所有可能构型进行全参数优化和频率计算,排除不存在的构型得到其优化构型。计算时对金属锰原子采用Hay[20]等人的含相对论校正的有效核电势价电子从头计算基组,即采用18-eECP双ξ基组(3s,3p,3d/2s,2p,2d),且P原子加极化函数ξP.d=0.55[21];所有计算均采用Gaussian09程序完成。
2 结果与讨论
2.1 各优化构型和能量
将原子簇Mn3BP分别构造成三角双锥、四方锥、平面五边形三种几何构型,变化不同原子所处位置,二、四重态各得到十种不同构型,再采用本文的模型和计算方法分别对其二、四重态构型进行优化计算。排除含有虚频的不稳定构型和几何构型相同的构型,最后得到四重态稳定构型五种,如图1 中1(4)~5(4)(右上角括号数字表示重态,下同),得到二重态稳定构型四种,如图1中1(2)~4(2),二、四重态构型分别按能量由低到高排序。
从图1中可以看出,各稳定构型的几何构型有戴帽三角锥(1(4)和4(4)),三角双锥(5(4)、1(2)和2(2)),平面四边形(3(4)和 3(2))和平面五边形(2(4)和4(2))。构型1(4)是以Mn1-Mn2-Mn3为基准面,B为锥顶,P为帽顶的戴帽三角锥;构型4(4)是以B-Mn1-Mn3为基准面,P为锥顶,Mn2为帽顶的戴帽三角锥。构型5(4)和 2(2)均是以 Mn1-Mn2-Mn3为基准面,P 为锥顶,B为锥底的三角双锥,优化构型相同,多重度不同;构型1(2)是以Mn1-Mn3-B为基准面,P为锥顶,Mn2为锥底的三角双锥。构型3(4)和3(2)均是以B原子作为中心原子,四边顶点以Mn1-Mn2-PMn3的排列顺序构成外形相似的平面四边形。构型2(4)和4(2)是由Mn1、Mn2、Mn3、B、P构成但排列顺序不同的平面五边形。

图1 团簇Mn3BP优化构型图Fig.1 Optimized configurations of cluster Mn3BP
表1列出了团簇Mn3BP各构型的总能量E、零点振动能EZP、校正能EZPE及其它能级参数,相同重态的各构型分别按能量由低到高排序。由表中数据知:整体上除构型1(2)外,四重态构型的能量均低于二重态的能量。分析各构型的总能量E、结合能 EBE、吉布斯自由能变 ΔG ,构型1(4)具有最低能量值(-343.050 a.u),最高结合能值(0.612 a.u)以及最高的吉布斯自由能变化值(-0.569 a.u),说明以Mn1-Mn2-Mn3为基准面,B为锥顶,P为帽顶的戴帽三角锥的构型1(4)是团簇Mn3BP各构型中最可能稳定存在的。构型 2(2)和 5(4)是相同的三角双锥,但重态不同,构型5(4)的能量值(-343.026 a.u)比构型2(2)的能量值(-343.018 a.u)低,构型5(4)的结合能值(0.589 a.u)比构型2(2)的结合能值(0.580 a.u)高,构型5(4)的吉布斯自由能变化的绝对值(0.544 a.u)比构型2(2)的吉布斯自由能变化的绝对值(0.536 a.u)高,说明相同几何构型的构型5(4)比构型 2(2)稳定;还有 3(2)和 3(4)的几何构型也几乎相同,重态不同,比较它们的各能级参数,四重态构型3(4)更可能稳定存在。以上几点说明多重度影响了相同构型的稳定性。比较表1所有二、四重态构型的各能级参数,可得结论:整体上除构型1(2)外,二重态构型的稳定性与四重态相比,四重态构型更为稳定。
为了更加系统地分析与比较各构型的稳定性大小,假定由稳定的原子Mn、B、P来形成团簇Mn3BP,即合成路线为:3Mn+B+P→Mn3BP。通过EBE=3EZPE(Mn)+EZPE(B)+EZPE(P)-EZPE(Mn3BP)计算各构型的结合能(EBE),通过ΔG=G(Mn3BP)-3 G(Mn)-G(B)-G(P)计算各构型的吉布斯自由能变化值(ΔG),结果见表1。为更直观分析各构型的EBE和ΔG的变化对构型稳定的影响,图2为各构型EBE和ΔG的变化趋势图。随着能量的增加,结合能(EBE)和吉布斯自由能变(ΔG)呈对称性变化,即它们的变化趋势相反。各稳定构型EBE逐渐减小,但均大于零;而ΔG逐渐增大,但均小于零。说明团簇Mn3BP各构型均能以合成路线:3Mn+B+P→Mn3BP自发结合形成,但自发程度不同。ΔG越大,自发程度越小;EBE越大,稳定性越好。因此,二、四重态各构型自发程度在不断减小,其中构型1(4)有最大的 EBE和最低 ΔG ,自发程度最大,稳定性最好;而构型4(2)与之恰好相反,自发程度最小,稳定性最差。
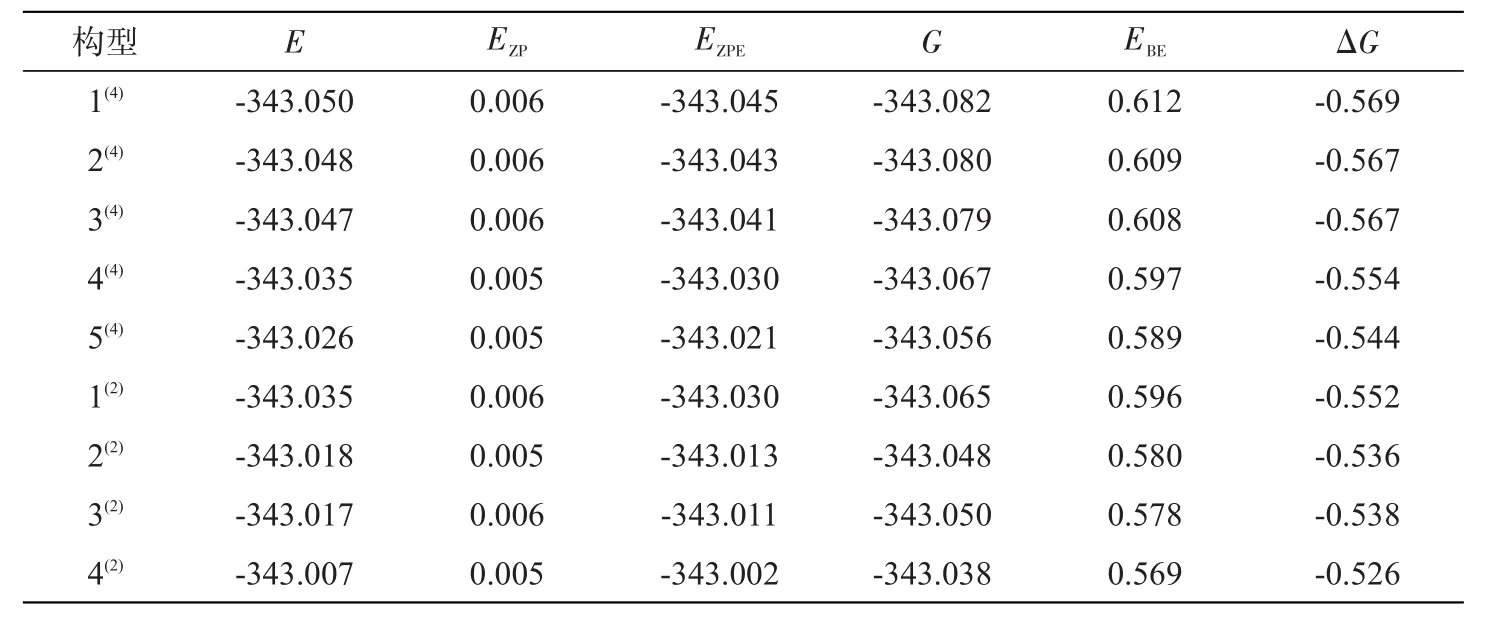
表1 团簇Mn3BP各构型能级参数,a.uTab.1 Energy level parameter for clusters Mn3BP,a.u
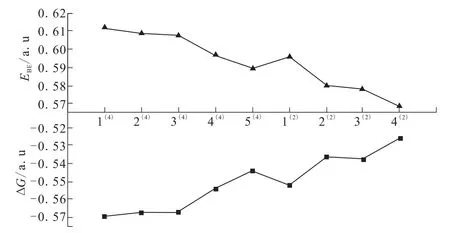
图2 团簇Mn3BP稳定构型的结合能EBE和吉布斯自由能变ΔG变化趋势图Fig.2 Variation trend of binding energy and Gibbs free energy change of each configuration in cluster Mn3BP
2.2 成键分析
表2列出了团簇Mn3BP各稳定构型的键长数据(Mn-P、Mn-B、Mn-Mn代表平均键长),图3是团簇Mn3BP的稳定构型的各平均键长变化趋势图。P-B键长范围为0.176~0.377 nm,变化幅度较大;Mn-P键长范围为0.244~0.376 nm,变化幅度较大;Mn-B键长范围为0.205~0.270 nm,变化幅度平缓;Mn-Mn键长范围为0.276~0.352 nm,变化幅度也比较平缓。在四重态构型中:Mn-P和Mn-B键在各构型中(除构型5(4)外)表现为协同作用,即同增同减;Mn-Mn和Mn-B键在各构型中表现为拮抗作用,即此消彼长。在二重态构型中:Mn-P和Mn-Mn键在各构型中(除4(2)外)中表现为协同作用,在构型4(2)中表现为拮抗作用;P-B和Mn-Mn键在各构型中表现为拮抗作用。
表3列出了团簇Mn3BP各稳定构型的键级数据(Mn-P、Mn-B、Mn-Mn代表平均键级),图4为团簇Mn3BP稳定构型的各键占总成键键级的百分比图。戴帽三角锥的构型 4(4),三角双锥的构型 2(2)和5(4),以及平面五边形的构型 4(2),这些构型中 P-B键的键级都为负值,占总成键键级比例为0,说明这些构型中P-B键对构型稳定性没有贡献或者贡献极小;Mn-Mn键在以上构型中占总成键键级比例最大不超过10%(9.76%),最小为0,而Mn-P键占总成键键级比例37.85%~51.34%,Mn-B键占总成键键级比例48.66%~52.59%,说明上述构型稳定性的主要贡献者是Mn-P键和Mn-B键。在其余构型中(1(4)、2(4)、3(4)、1(2)、3(2))P-B 键占总成键
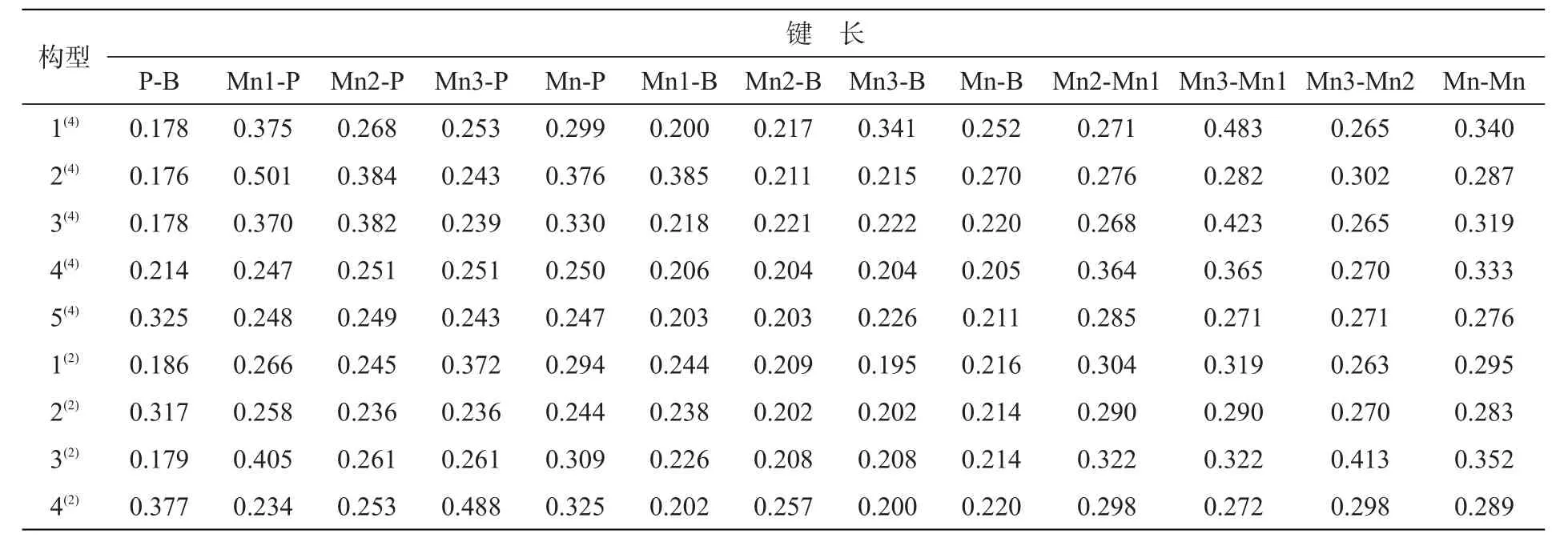
表2 团簇Mn3BP稳定构型的键长,nmTab.2 Bond length of cluster Mn3BP,nm
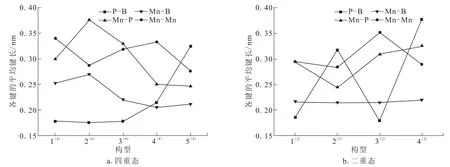
图3 团簇Mn3BP各稳定构型的平均键长趋势图Fig.3 Average bond length in stable configurations in cluster Mn3BP
键级比例16.85%~29.58%,Mn-P键占总成键键级比例17.77%~28.07%,Mn-B键占总成键键级比例32.12%~47.91%,Mn-Mn键占总成键键级比例10.65%~21.76%,说明各键对上述构型稳定性都有贡献,其中Mn-B键贡献最大,Mn-Mn键贡献最小。综上所述,团簇Mn3BP各稳定构型中,金属与非金属成键的Mn-P和Mn-B键是构型稳定的主要贡献者。
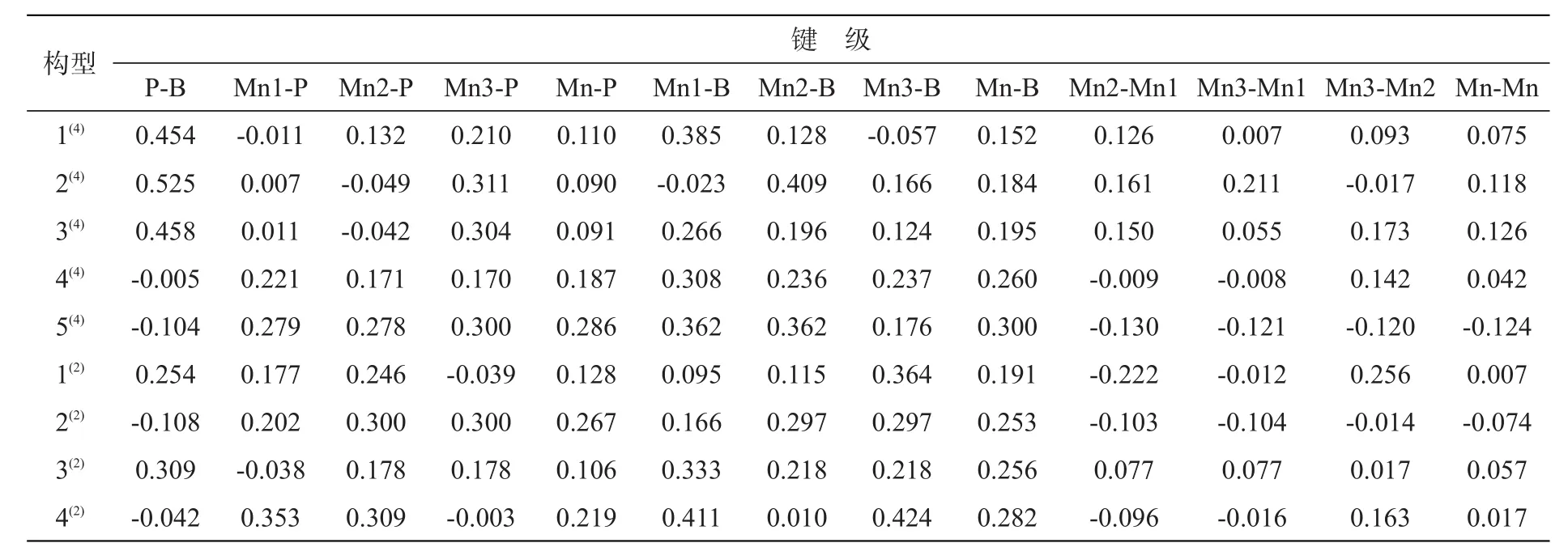
表3 团簇Mn3BP稳定构型的键级Tab.3 Mulliken bond order of cluster Mn3BP
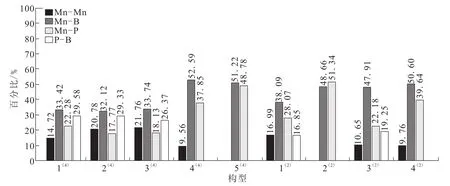
图4 团簇Mn3BP稳定构型的各键占总成键键级的百分比,%Fig.4 Percentage of total bond order of each stableconfiguration in cluster Mn3BP,%
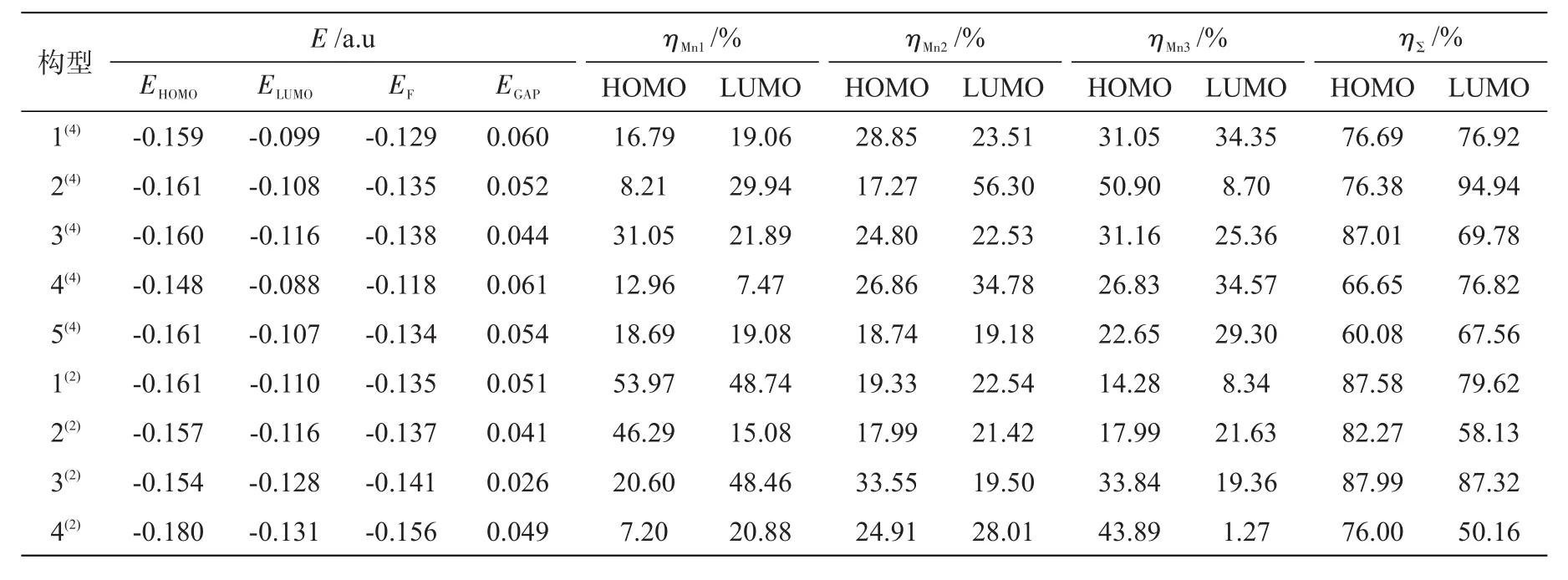
表4 团簇Mn3BP的能级参数和Mn原子的3d轨道在HOMO轨道和LUMO轨道上的贡献Tab.4 Parameters of energy level,contribution of Mn atoms to 3d orbitals in HOMO and LUMO of cluster Mn3BP
2.3 各优化构型的HOMO和LUMO轨道的贡献
表4列出了团簇Mn3BP各构型的部分能级参数及每个Mn原子的3d轨道在HOMO轨道和LUMO轨道上的贡献数据,ηMn代表Mn原子的贡献率,ηΣ代表所有Mn原子的贡献率,图5是其总贡献率图。在所有构型中,Mn原子在最高占据轨道(HOMO)的总贡献率为60.08%~87.99%,在最低未占据轨道(LUMO)的总贡献率为50.16%~94.94%,都大于50%,由此可知Mn3BP团簇潜在的活性位点是金属原子Mn,并且其前线轨道的主要贡献者也是Mn原子。构型3(2)的能隙差EGAP=0.026 a.u,在所有构型中最小,说明构型3(2)最为活泼,反应的活性最好;构型4(4)的能隙差 EGAP=0.061 a.u,在所有构型中最大,说明该构型不易发生电子转移跃迁,相对其余构型不易发生化学反应。
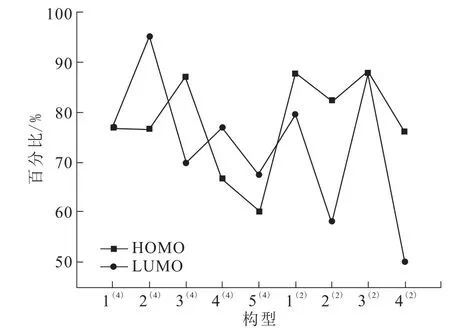
图5 团簇Mn3BP稳定构型Mn原子的3d轨道在HOMO和LUMO轨道上的贡献率,%Fig.5 Contribution rateof 3d orbitals of Mn atoms to HOMO and LUMO of cluster Mn3BP,%
3 结论
团簇Mn3BP的优化构型共有9种,分别为:戴帽三角锥(1(4)和 4(4)),三角双锥(1(2)、2(2)和 5(4)),平面四边形(3(2)和 3(4))和平面五边形(4(2)和2(4));整体上除构型1(2)外,二重态构型的稳定性没有四重态构型好,多重度是影响构型稳定的因素之一;在所有构型中,构型1(4)最为稳定,构型4(2)最不稳定;四重态构型中Mn-P和Mn-B键之间主要存在协同作用,Mn-Mn和Mn-B键之间主要存在拮抗作用;在二重态构型中Mn-P和Mn-Mn键之间主要存在协同作用,P-B和Mn-Mn键之间主要存在拮抗作用;各稳定构型中,Mn-P键和Mn-B键是构型稳定的主要贡献者;各构型前线轨道的主要贡献者是Mn原子,并且各构型的潜在活性位点也是Mn原子;在所有构型中,构型3(2)的化学性质最活泼,构型4(4)的最不易发生化学反应。
[1]SOMMER R L,CHIEN C L.Role of magnetic anisotropy in the magnetoimpedance effect in amorphous alloys[J].Applied Physics Letters,1995,67(6):857-859.
[2]EGAMI T.Low-field magnetic properties of amorphous alloys[J].Journal of the American Ceramic Society,1977,60(3-4):128-133.
[3]ZHANG W,INOUE A.Crystallization and hard magnetic properties of Fe-Co-Nd-Dy-B amorphous alloys with glass transition[J].Journal of Applied Physics,2000,87(9):6122-6124.
[4]GREER A L,RUTHERFORD K L,HUTCHINGS I M.Wear resistance of amorphous alloys and related materials[J].International Materials Reviews,2002,47(2):87-112..
[5]SOUZA C A C D,BOLFARINI C,BOTTA W J,et al.Corrosion resistance and glass forming ability of Fe47Co7Cr15M9Si5B15Y2(M=Mo,Nb) amorphous alloys[J].Materials Research,2013,16(6):1294-1298.
[6]KIM Y H,HIRAGA K,INOUE A,et al.Crystallization and high mechanical strength of Al-based amorphous alloys[J].Materials Transactions Jim,1994,35(5):293-302.
[7]DENG J F,LI H,WANG W.Progress in design of new amorphous alloy catalysts[J].Catalysis today,1999,51(1):113-125.
[8]FANG Z G,GUO J X.Theoretical study on the catalytic activity and sulfur resistibility of amorphous alloy Ni-B-P[J].结构化学,2007,26(3):273-280.
[9]LI H,WANG Y,ZHAO Q,et al.Ultrasound-assisted synthesis of monodisperse Ru-B amorphous alloys with enhanced catalytic activity in maltose hydrogenation[J].Research on Chemical Intermediates,2009,35(6-7):779-790.
[10]SZUMMER A,JANIKCZACHOR M,MOLNÁR Á,et al.Hydrogenation under high pressure enhancing catalytic activity of Cu-Zr amorphous alloys[J].Journal of Physics Condensed Matter,2002,14(141):11405-11409.
[11]BEDNARSKA L M,HERTSYK O M,KOVBUZ M O,et al.Influence of the elemental composition of the amorphous metallic alloys Fe-Me-Si-B on the catalytic activity in the red/ox reactions[J].Physics and Chemistry of Solid State,2007,8(3):533-537.
[12]CHUI Z J,WANG L,WANG K Y,et al.The catalytic properties of Fe78Si12B10,alloys prepared by ball milling[J].Materials Science&Engineering A,1991,134(91):1037-1040.
[13]ZHANG J,ZHU R.Thermodynamic properties of Mn-P and Fe-Mn-P melts[J].矿物冶金与材料学报,2000,7(1):10-13.
[14]SINHA A K,DUWEZ P.Radial distribution function of an amorphous Mn-P-C alloy[J].Journal of Applied Physics,1972,43(2):431-434.
[15]WOLANSKA N,LIS A K,LIS J.Investigation of CMn-B steel after hot deformation[J].Archives of Materials Science&Engineering,2007,28(2):119-125.
[16]ORIO M,PANTAZIS D A,NEESE F.Density functional theory[J].Photosynthesis research,2009,102(2-3):443-453.
[17]CAR R,Parrinello M.Unified approach for molecular dynamics and density-functional theory[J].Physical review letters,1985,55(22):2471-2474.
[18]CRAMER C J,TRUHLAR D G.Density functional theory for transition metals and transition metal chemistry[J].Physical Chemistry Chemical Physics,2009,11(46):10757-10816.
[19]YU X.Hyperbolic multi-topology and the basic principle in quantum mechanics[J].Advances in Applied Clifford Algebras,1999,9(1):109-118.
[20]HAY P J,WADT W R.Ab initio effective core potentials for molecular calculations.Potentials for K to Au including the outermost core orbitals[J].The Journal of Chemical Physics,1985,82(1):299-310.
[21]FANG Z G,HU H Z,GUO J X.Quantum chemical study on geometry and property of cluster Ni4P[J].结构化学,2006,25(1):7-16.
猜你喜欢
辽宁科技大学学报(2022年5期)2023-01-04
山西大学学报(自然科学版)(2022年5期)2022-11-23
大学物理(2022年9期)2022-09-28
物理通报(2020年7期)2020-07-01
原子与分子物理学报(2020年5期)2020-03-17
考试周刊(2018年39期)2018-04-19
原子与分子物理学报(2015年3期)2015-11-24
学园(2015年5期)2015-10-21
海外文摘(2001年1期)2001-04-09
