
过敏毒素C3a在健康人气道上皮间质转化中的作用研究*
2018-01-05 05:38刘代顺吴凯峰丁宏伟刘建英
重庆医学 2017年34期
李 竹,刘代顺,吴凯峰,丁宏伟,刘建英△
(1.遵义医学院第三附属医院呼吸内科,贵州遵义 563000;2.遵义医学院公共卫生学院,贵州遵义 563000)
过敏毒素C3a在健康人气道上皮间质转化中的作用研究*
李 竹1,刘代顺1,吴凯峰1,丁宏伟2,刘建英1△
(1.遵义医学院第三附属医院呼吸内科,贵州遵义 563000;2.遵义医学院公共卫生学院,贵州遵义 563000)
目的观察过敏毒素C3a在正常人气道上皮间质转化(EMT)中的作用及其相关分子机制。方法培养正常人气道上皮细胞BEAS-2B,分为对照组,重组人(rhC3a)刺激组,rhC3a+C3a受体拮抗剂(C3aRA)的拮抗组,通过显微镜观察细胞形态学变化情况;四甲基偶氮唑蓝(MTT)法检测细胞增殖能力;ELISA检测细胞上清液中转化生长因子-β1(TGF-β1)的蛋白表达情况;RT-PCR检测C3a受体(C3aR)和EMT相关指标mRNA变化情况;Western blot检测C3aR、Smad2/3、p38-MAPK蛋白表达情况。结果30、50 nmol/L rhC3a刺激组细胞形态由正常鹅卵石样变为梭形,加入1 μmol/L C3aRA拮抗组细胞形态较对照组比较无明显改变。50 nmol/L rhC3a刺激组的细胞增殖较对照组降低(P=0.047);30 nmol/L rhC3a刺激组中TGF-β1蛋白水平较对照组升高(P<0.05),C3aR mRNA及蛋白水平较对照组均升高(P<0.05),p-Smad2/3、p-p38-MAPK蛋白水平较对照组升高(P<0.05),加入1 μmol/L C3aRA可以降低以上指标表达(P<0.05)。30 nmol/L rhC3a刺激组中α-平滑肌肌动蛋白、N-钙黏蛋白mRNA表达上调,E-钙黏蛋白mRNA表达下降,加入1 μmol/L C3aRA可以拮抗这一过程(P<0.05)。结论过敏毒素C3a通过结合C3aR,诱导正常人气道上皮细胞发生EMT,其机制可能是通过激活Smad2/3、p38-MAPK通路来参与。
气道重塑;纤维化;过敏毒素C3a;上皮间质转化
慢性阻塞性肺疾病(chronic obstructive pulmonary disease,COPD)是可治疗和可预防的一种限制性的不可逆气流受限的一组慢性肺部疾病。COPD发病机制复杂,气流受限通常是呈进行性发展,小气道的纤维化和闭塞可能是导致呼吸道生理功能障碍的主要原因。近年来研究发现,在COPD患者气道中,尤其是吸烟者的气道上皮间质转化(epithelial mesenchymal transition,EMT)是活跃的。EMT是上皮细胞逐渐转化为间充质样细胞并失去其上皮细胞的功能和特性的过程,被认为参与气道重塑[1]。补体系统参与机体的特异性和非特异性免疫机制,表现为抗微生物防御反应,免疫调节及介导免疫病理的损伤性反应,其中补体C3是补体系统中含量最高的成分,C3a是补体C3活化过程中产生的裂解片段,具有过敏毒素作用,被报道参与气道炎症过程[2]。最新研究表明,C3a除具有促炎作用外,有可能是一种新的促纤维化因子[3]。有报道证实,C3a能够促进肺纤维化,与此同时阻断C3a与其受体C3aR的结合,可以阻止肺纤维化的进展[4]。但是,C3a是否与气道EMT有关目前尚无报道。本课题旨在探讨过敏毒素C3a在正常人气道EMT中的作用及其相关分子机制,期待为预防COPD气道重塑提供新的靶点。
1 材料与方法
1.1材料
1.1.1实验细胞与试剂 正常人气道上皮细胞BEAS-2B(美国ATCC公司),重组人C3a(rhC3a),ELISA试剂盒(美国R&D公司),C3aR拮抗剂(C3aRA/SB290157,艾美捷科技公司),DMEM F12培养基(美国Hyclon公司),胎牛血清(杭州四季青公司),四甲基偶氮唑蓝青链霉素(北京索莱宝公司),RT-PCR试剂盒(日本TaKaRa公司),兔抗-GAPDH一抗、兔抗-C3aR、山羊抗兔-IgG/HRP、兔抗-p-Smad2/Smad3一抗、兔抗-p-p38-MAPK一抗(北京博奥森公司),兔抗-Smad2/Smad3、兔抗-MAPK14/P38一抗(武汉博士德公司)。
1.1.2实验仪器 生物安全柜、ND1000核酸蛋白测定仪(美国Thermo 公司),光学显微镜(日本Olympus公司),荧光显微镜(德国Leica公司),低温高速离心机、移液器(德国Eppendorf 公司),Stax Fax2100 型酶标仪(美国Awarn-er公司),电泳槽、电泳仪、凝胶成像仪(美国Bio-Rad公司)。
1.2方法
1.2.1细胞培养及形态观察 BEAS-2B细胞用含5%胎牛血清的DMEM F12培养基,置于37 ℃、5% CO2培养箱中培养,细胞密度达70%~80%传代,取对数生长期细胞进行实验。分为3组,培养细胞0~72 h后观察每组细胞形态变化情况。
1.2.2四甲基偶氮唑蓝(MTT)细胞增殖实验 以每组每孔1×103个细胞接种于96孔板,加入5% DMEM F12培养24 h,倒掉原培养基,加入无血清培养基饥饿6 h,弃去培养基,加入不同浓度重组人C3a刺激,在96孔板周围一圈加入磷酸盐缓冲液(PBS),分别培养1、24、48、72 h。吸去上清液,加入90 μL 5% DMEM F12继续培养4 h,然后吸去上清液,每孔加入110 μL Formazan溶解液,置摇床上低速震荡10 min,使结晶物充分溶解,在ELISA检测仪490 nm处测量各孔的吸光度(A)值。每组设置3个复孔,取平均值,绘制生长曲线,实验重复3次。
1.2.3ELISA检测细胞上清液中转化生长因子-β1(TGF-β1)的蛋白水平 将收集好的细胞上清液从-80 ℃冰箱取出置于冰盒上,在酶标包被板上设标准孔10孔,加样,分别设空白孔、待测样品孔,用封板膜封板后置37 ℃温育30 min,洗涤5次,拍干,每孔加入酶标试剂50 μL,空白孔除外,再次温育30 min(条件同前),洗涤5次,拍干,加入显色剂,37 ℃避光显色15 min,每孔加入终止液50 μL。在15 min以内,ELISA检测仪450 nm处测量各孔的A值。
1.2.4RT-PCR检测不同基因mRNA的表达 不同组细胞待细胞密度达80%时,加入1 mL Trizol提取液提取细胞总RNA,具体方法按照TaKaRa RT-PCR试剂盒说明书操作。PCR引物由Invitrogen和上海生物工程公司合成,具体序列见表1。
1.2.5蛋白免疫印迹(Western blot)检测不同蛋白水平 以每组每孔1×106个细胞接种于6孔板,待细胞密度达80%时,加入RIPA裂解液提取蛋白,根据二喹啉甲酸(BCA)蛋白定量试剂盒说明书测定蛋白水平。30 μg总蛋白在10%十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)分离后,迁移到聚偏氟乙烯(PVDF)膜,5%脱脂奶粉4 ℃摇床封闭2 h,洗膜,加入稀释好的相应一抗4 ℃孵育过夜(不超过18 h),洗膜,加入稀释好的二抗37 ℃孵育2 h,电化学发光(ECL)试剂盒检测。


表1 PCR基因引物序列
2 结 果
2.1rhC3a诱导BEAS-2B细胞发生形态改变 正常BEAS-2B细胞形态呈鹅卵石或铺路石样,分别加入10、30、50 nmol/L rhC3a刺激细胞0、24、48、72 h后观察每组细胞形态变化情况。观察到30、50 nmol/L组细胞在48、72 h,细胞由正常形态变为梭形或长条形,见图1。
2.2C3aRA可以抑制BEAS-2B细胞发生形态改变 分别加入30、50 nmol/L rhC3a刺激细胞30 min后,加入1 μmol/L C3aRA,观察到在48、72 h拮抗组细胞形态较刺激组未发生明显条形或梭形变,见图2。
2.350 nmol/L rhC3a降低BEAS-2B细胞的增殖 分别加入0、10、30、50 nmol/L rhC3a刺激BEAS-2B细胞1、24、48、72 h,观察每组细胞的增殖情况。发现50 nmol/L rhC3a组在72 h较对照组和10 nmol/L rhC3a组比较,增殖下降,见表2。
2.4rhC3a可以诱导TGF-β1水平的激活 用ELASA法检测各组细胞上清液中TGF-β1的蛋白水平结果发现,在不同时间点(12、24、48、72 h),30 nmol/L rhC3a组中TGF-β1蛋白水平均显著高于对照组和10 nmol/L rhC3a组,加入1 μmol/L C3aRA均可以抑制TGF-β1的蛋白水平,见表3。
2.5rhC3a可诱导C3aR活化,C3aRA能够抑制C3aR的活化用RT-PCR法检测C3aR的mRNA水平,结果发现,在48 h,30 nmol/L rhC3a组中C3aR的mRNA水平显著高于对照组和10 nmol/L rhC3a组,加入1 μmol/L C3aRA可以抑制C3aR mRNA的水平,其中对照组(0 nmol/L rhC3a)、1 μmol/L C3aRA组、10 nmol/L rhC3a组、10 nmol/L rhC3a+1 μmol/L C3aRA组、30 nmol/L rhC3a组、30 nmol/L rhC3a+1 μmol/L C3aRA组中C3aR mRNA水平分别为0.085±0.018、0.078±0.025、0.065±0.020、0.097±0.029、0.338±0.031、0.092±0.017,见图3。 另外,用Western blot法检测 C3aR的蛋白水平,得到同样结果,30 nmol/L rhC3a刺激细胞48 h后,C3aR的蛋白水平显著高于对照组,加入1 μmol/L C3aRA可以抑制C3aR的蛋白水平,其中对照组(0 nmol/L rhC3a)、30 nmol/L rhC3a组、30 nmol/L rhC3a+1 μmol/L C3aRA组的C3aR mRNA表达水平分别为0.835±0.003、1.381±0.016、1.315±0.007,见图4。
2.6rhC3a可诱导BEAS-2B细胞发生EMT,C3aRA能够抑制EMT过程 分为对照组(0 nmol/L rhC3a)、1 μmol/L C3aRA组、10 nmol/L rhC3a组、10 nmol/L rhC3a+1 μmol/L C3aRA组、30 nmol/L rhC3a组、30 nmol/L rhC3a+1 μmol/L C3aRA组,作用48、72 h,用RT-PCR法检测EMT相关指标的mRNA水平。结果发现,10 nmol/L、30 nmol/L rhC3a组细胞48 h和72 h后,α平滑肌肌动蛋白(α-SMA)mRNA水平较对照组和1 μmol/L C3aRA组升高,加入受体拮抗剂后可以抑制其mRNA水平的表达;在48 h,仅30 nmol/L rhC3a组N-钙黏蛋白(N-cadherin)mRNA水平高于对照组,加入受体拮抗剂后可以抑制其mRNA水平的表达,在72 h,10 nmol/L、30 nmol/L rhC3a组N-cadherin mRNA水平均高于对照组,加入受体拮抗剂同样可以抑制其mRNA水平的表达,且1 μmol/L C3aRA组N-cadherin的mRNA水平低于对照组;在30 nmol/L rhC3a刺激48、72 h后,E-钙黏蛋白(E-cadherin)的mRNA水平较对照组降低,加入受体拮抗剂后其mRNA水平升高,见图5,表4、5。
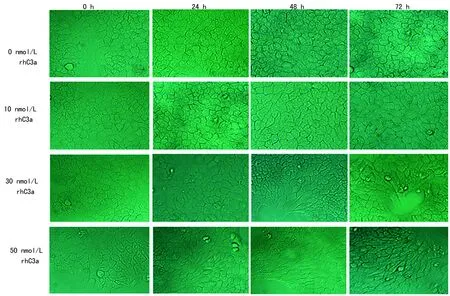
图1 不同浓度rhC3a刺激BEAS-2B细胞后的形态变化情况(×200)表2 不同浓度rhC3a作用下BEAS-2B细胞的A值
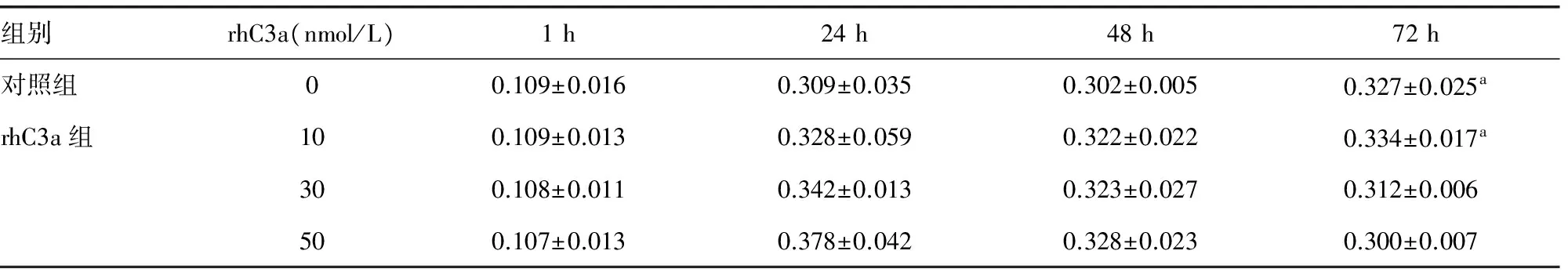
组别rhC3a(nmol/L)1h24h48h72h对照组00.109±0.0160.309±0.0350.302±0.0050.327±0.025arhC3a组100.109±0.0130.328±0.0590.322±0.0220.334±0.017a300.108±0.0110.342±0.0130.323±0.0270.312±0.006500.107±0.0130.378±0.0420.328±0.0230.300±0.007
a:P<0.05,与50 nmol/L rhC3a组比较

表3 不同浓度rhC3a联合C3aRA对TGF-β1蛋白水平的影响
续表3 不同浓度rhC3a联合C3aRA对TGF-β1蛋白水平的影响
a:P<0.05,各组与30 nmol/L rhC3a+1 nmol/L C3aRA组比较

图2 不同组BEAS-2B细胞的形态变化情况(×200)

图3 rh C3a和C3aRA对C3aR mRNA水平的影响

图4 rhC3a和C3aRA对C3aR 蛋白水平的影响
2.7rhC3a对Smad2/3、p38-MAPK蛋白表达的影响 采用30 nmol/L rhC3a刺激BEAS-2B细胞48 h,用Western blot检测Smad2/3、p38-MAPK的蛋白表达水平。结果发现,30 nmol/L rhC3a组的p-Smad2/3、p-p38-MAPK蛋白水平较对照组(0 nmol/rhC3a)增加,加入受体拮抗剂可以抑制其蛋白的表达,而总Smad2/3(P=0.451)、总p38-MAPK(P=0.263)的蛋白水平与对照组比较差异无统计学意义,见图6、表6。

A:作用48 h;B:作用72 h

图5 rhC3a和C3aRA对EMT相关指标 mRNA水平的影响表4 不同浓度rhC3a联合C3aRA作用48 h对EMT相关指标mRNA水平的影响
表5 不同浓度rhC3a联合C3aRA作用72 h对EMT相关指标mRNA水平的影响
表6 rhC3a、C3aRA对Smad2/3、p-p38-MAPK蛋白及其磷酸化水平的影响

图6 rhC3a对Smad2/3、p38 MAPK通路的影响
3 讨 论
COPD的主要特征在于小气道重塑与细支气管周围纤维化及肺气肿,其中,以小气道纤维化尤为重要[5]。由于持续性的气流受限,COPD患者的肺功能呈进行性减退,严重影响其劳动力和生活质量,并造成巨大的社会和经济负担。根据世界卫生组织统计,在20世纪有1亿人因COPD死亡[6],2014年发布的数据表明,至2030年COPD将上升成为全球第三大死亡原因[7]。
目前认为EMT深入地参与了COPD的病理学改变。它涉及气道的纤维化和气流阻塞,也可能与COPD患者肺癌的高患病率有关[8]。EMT通过气道上皮细胞的活化变成成纤维细胞,而成纤维细胞通过刺激上皮细胞来刺激产生更多的EMT,从而促进小气道狭窄和气流阻塞的恶性循环。从COPD患者气道活检中证实了各种蛋白质的表达上调,成为EMT的生物标记物。如α-SMA,波形蛋白(vimentin)和纤维连接蛋白(fibronectin),负责组织重塑和纤维化[9],参与间充质细胞蛋白的表达。E-cadherin的表达下降,被认为是EMT上皮细胞的标记物,同时伴随N-cadherin的表达增加,这是间充质细胞的标记物,这种变化通常被称为“钙黏蛋白的转换”[10]。
C3a已被证实是一种促炎因子,在COPD患者的诱导痰和血清中表达均增加[11]。在近年来的研究中表明,其的确有促纤维化作用,除了参与肺纤维化的过程以外,在肾脏疾病中,C3a也可诱导肾小管上皮细胞发生EMT,通过C3aRA可以减弱TGF-β/Smad3诱导的肾小管EMT[12-13]。目前,暂无C3a在气道EMT中的作用研究,但已有研究证实,促纤维化密切相关的因子TGF-β1 能够促进气道EMT的发生[14]。因此本研究采用rhC3a刺激正常人气道上皮细胞,发现TGF-β1水平的激活,并证实了C3aR水平的活化,以及EMT的发生,而C3aRA则能够抑制这一过程,猜测rhC3a可能是EMT过程的上游信号分子。C3aRA是C3aR的拮抗剂,通常拮抗剂阻断的是配体和受体的结合,而在本研究中能够抑制受体本身的水平,其中的具体机制尚不清楚[15]。 除此之外,有研究表明在人类肺泡和支气管上皮细胞系中,TGF-β1可以通过激活Smad2诱导EMT的发生[16]。尿激酶型纤溶酶原激活物受体(uPAR)信号通路通过激活许多细胞信号因子能够诱导EMT的发生,包括磷脂酰肌醇3-激酶(PI3k)、Src家族激酶、Akt、ERK/MAPK等[17]。而本实验通过研究C3a与Smad2/3、p38-MAPK信号通路的关系,首次证实了C3a可以激活Smad2/3、p38-MAPK信号通路。
综上所述,C3a通过结合C3aR,可诱导正常人气道上皮细胞发生EMT,其机制可能是通过激活Smad2/3、p38-MAPK通路来参与。本课题组今后也将继续深入研究其可能的机制并对相关动物模型进行探讨。C3a有望成为气道重塑干预治疗的新靶点,对COPD的治疗和预后有着重大意义。
[1]Bartis D,Bartis D,Mise N,et al.Epithelial-mesenchymal transition in lung development and disease:does it exist and is it important?[J].Thorax,2014,69(8):760-765.
[2]Marc MM,Korosec P,Kosnik M,et al.Complement factors C3a,C4a,and C5a in chronic obstructive pulmonary disease and asthma[J].Am J Respir Cell Mol Biol,2004,31(2/1):216-219.
[3]Tang Z,Lu B,Hatch E,et al.C3a mediates epithelial-to-mesenchymal transition in proteinuric nephropathy[J].J Am Soc Nephrol,2009,20(3):593-603.
[4]Gu HM,Fisher AJ,Mickler EA,et al.Contribution of the anaphylatoxin receptors,C3aR and C5aR,to the pathogenesis of pulmonary fibrosis[J].FASEB J,2016,30(6):2336-2350.
[5]Hogg JC.Pathophysiology of airflow limitation in chronic obstructive pulmonary disease[J].Lancet,2004,364(9435):709-721.
[6]Laniado-Laborín R.Smoking and chronic obstructive pulmonary disease(COPD).Parallel epidemics of the 21 century[J].Int J Environ Res Public Health,2009,6(1):209-224.
[7]Copd BO.WHO.Burden of COPD[EB/OL].[2017-01-01].http://www.who.int/respiratory/copd/burden/en/,2016.
[8]Nowrin K,Sohal SS,Peterson GA,et al.Epithelial-mesenchymal transition as a fundamental underlying pathogenic process in COPD airways:fibrosis,remodeling and cancer[J].Expert Rev Respir Med,2014,8(5):547-559.
[9]Günther A,Korfei M,Mahavadi P,et al.Unravelling the progressive pathophysiology of idiopathic pulmonary fibrosis[J].Eur Respir Rev,2012,21(124):152-160.
[10]Zeisberg M,Neilson EG.Biomarkers for epithelial-mesenchymal transitions[J].J Clin Invest,2009,119(6):1429-1437.
[11]Zhang J,Yao WZ,Chen YH.Change in airway anaphylatoxin-complement factors C3a of sputum in patients with chronic obstructive pulmonary disease[J].Beijing Da Xue Xue Bao,2011,43(3):446-449.
[12]Li L,Yin QH,Tang X,et al.C3a receptor antagonist ameliorates inflammatory and fibrotic signals in type 2 diabetic nephropathy by suppressing the activation of TGF-beta/smad3 and IKB alpha pathway[J].PLoS One,2014,9(11):e113639.
[13]Li L,Chen LJ,Zang J,et al.C3a and C5a receptor antagonists ameliorate endothelial-myofibroblast transition via the Wnt/beta-catenin signaling pathway in diabetic kidney disease[J].Metabolism,2015,64(5):597-610.
[14]Gohy ST,Hupin C,Fregimilicka C,et al.Imprinting of the COPD airway epithelium for dedifferentiation and mesenchymal transition[J].Eur Respir J,2015,45(5):1258-1272.
[15]Lim J,Iyer A,Suen JY,et al.C5aR and C3aR antagonists each inhibit diet-induced obesity,metabolic dysfunction,and adipocyte and macrophage signaling[J].FASEB J,2013,27(2):822-831.
[16]Xu J,Lamouille S,Derynck R.TGF-beta-induced epithelial to mesenchymal transition[J].Cell Res,2009,19(2):156-172.
[17]Lester RD,Jo M,Montel V,et al.uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells[J].J Cell Biol,2007,178(3):425-436.
StudyontheroleofallergictoxinC3ainepithelialmesenchymaltransitionofnormalhumanbronchialepitaheliumcells*
LiZhu1,LiuDaishun1,WuKaifeng1,DingHongwei2,LiuJianying1△
(1.DepartmentofRespiratoryMedicine,ThirdAffiliatedHospitalofZunyiMedicalCollege,Zunyi,Guizhou563000,China;2.SchoolofPublicHealth,ZunyiMedicalCollege,Zunyi,Guizhou563000,China)
ObjectiveTo investigate the role of anaphylatoxin C3a on epithelial mesenchymal transition(MTT) in normal human bronchial epithelium cells and its molecular mechanism.MethodsNormal human bronchial epithelium cells BEAS-2B were cultured,and divided into control group,rhC3a stimulation group,rhC3a+C3a receptor(C3aRA)antagonist group,the morphological changes of cells were observed by microscope;cell proliferation was detected by MTT;the expression of TGF-β1 protein level in cell supernatant was evaluated by ELISA;the expression of C3aR mRNA and EMT related indicators mRNA changes were detected by RT-PCR;the expression of C3aR and Smad2/3,p38 MAPK pathway proteins were detected by Western blot.ResultsCell morphology in 30,50 nmol/L rhC3a stimulation group was changed from normal cobblestone like to spindle shape,cell morphology in C3aRA antagonist group had no significant change when compared with the control group.The cell proliferation was reduced in 50 nmol/L rhC3a stimulation group(P=0.047);the levels of TGF-β1,p-Smad2/3,p-p38-MAPK protein were increased(P<0.05),C3aR mRNA and protein levels were also significantly increased (P<0.05) in 30 nmol/L rhC3a stimulation group when compared with control group,but the addition of 1 μmol/L C3aRA could reduce their expressions(P<0.05).30 nmol/L rhC3a could induce the up regulation of α -SMA and N-cadherin mRNA,and decrease the expression of E-cadherin mRNA,adding 1 μmol/L C3aRA can antagonize this process (P<0.05).ConclusionAnaphylatoxin C3a can induce EMT in normal human bronchial epithelium cells by combining C3aR,its mechanism may be involved in activating Smad2/3 and p38-MAPK pathway.
airway remodeling;fibrosis;anaphylatoxin C3a;epithelial mesenchymal transition
贵州省科学技术基金(黔科合J字[2013]2310号)。
李竹(1989-),住院医师,硕士,主要从事慢性阻塞性肺疾病方面的研究。△
,E-mail:ljy2317@126.com。
10.3969/j.issn.1671-8348.2017.34.002
R714.253
A
1671-8348(2017)34-4757-06
2017-08-21
2017-09-29)
猜你喜欢
传染病信息(2022年3期)2022-07-15
国际呼吸杂志(2019年22期)2019-12-09
国际呼吸杂志(2019年5期)2019-03-30
国际呼吸杂志(2019年3期)2019-03-01
国际呼吸杂志(2019年2期)2019-02-14
海南医学(2016年8期)2016-06-08
中华老年多器官疾病杂志(2016年7期)2016-04-28
中国社区医师(2015年14期)2015-12-24
医学研究杂志(2015年6期)2015-07-01
中华皮肤科杂志(2014年4期)2014-12-19
