
两亲性介孔碳材料负载Pt催化硝基苯制对氨基苯酚
2018-01-02 06:42梁继芬郝雅娟杨恒权
山西大学学报(自然科学版) 2017年4期
梁继芬,郝雅娟,杨恒权*
(1.山西大学 分子科学研究所,山西 太原 030006;2.山西大学 化学化工学院,山西 太原 030006)
两亲性介孔碳材料负载Pt催化硝基苯制对氨基苯酚
梁继芬1,郝雅娟2,杨恒权2*
(1.山西大学 分子科学研究所,山西 太原 030006;2.山西大学 化学化工学院,山西 太原 030006)
以氟碳表面活性剂(FC4)和Pluronic F127在酸性乙醇溶液中形成的胶束作为模板制备具有明显的介孔孔道结构的聚合物(RF),然后以400oC碳化后的材料(C-400)为载体,采用浸渍法负载Pt制备得到催化剂Pt/C-400。通过扫描电子显微镜(SEM)、透射电子显微镜(TEM)、N2吸附-脱附对载体和催化剂的结构进行表征,并采用油-水双相体系对其亲疏水性测试。研究表明,制备的Pt/C-400金属颗粒分散均匀,孔径尺寸均一,介孔孔道结构明显,具有一定的表面两亲性。该催化剂在硝基苯催化加氢生成对氨基苯酚反应中,硝基苯的转化率和对氨基苯酚的选择性可分别达到94 %和76 %,中间体苯基羟胺(PHA)的选择性明显高于负载量为5 %的商业化Pt/C催化剂,表明两亲性表面催化剂有助于中间体从活性位点的脱附,在一定程度上降低了反应传质阻力,从而提高了反应选择性。
两亲性介孔碳材料;硝基苯加氢;Bamberger重排;氟碳表面活性剂
0 引言
介孔碳因其密度小、比表面积大和卓越的吸附性能一直吸引着科学家的关注。但由于介孔碳的孔道尺寸分布广、孔道结构不可控在很大程度上限制了它的应用开发[1]。科研工作者们长期以来致力于有序介孔碳材料的制备和性能研究,以介孔硅作为硬模板制备介孔碳材料,可通过调控模板硅的孔道结构实现碳材料的结构调控,但由于过程复杂,孔径、形貌调控有限以及碳源和模板硅间的相互作用力弱等因素极大地限制了碳材料的进一步发展[2]。为了解决上述问题,国内外学者经过探索与优化,提出了以表面活性剂形成的胶束与碳前驱体进行有机-有机自组装制备有序介孔材料的软模板法。该方法制备的碳材料孔隙有序,孔径分布集中,可以形成如Ia3d,P6mm,Im3m,Fd3m 和Fm3m等结构[3-6]。受Stöber法合成二氧化硅球的启发,刘健等人将改进的Stöber法应用于介孔碳材料的制备,以间苯二酚和甲醛作为碳源,碱性(NH3·H2O)条件下制备出了粒径均一(~520 nm)、分散性好的纳米碳球[7]。随后的研究发现,表面活性剂的选择对材料孔道的构造起着至关重要的作用,并利用P123、F127、CTAB、FC4等不同种类的表面活性剂,实现了对碳材料粒径、孔道结构的调控[6,8-10]。随着尺寸、结构可控碳材料制备方法的日益成熟,碳材料的性能研究和应用开发发展迅速,在催化、吸附、净化、能源存储和转化等领域发挥着越来越重要的作用[2,11-13],而材料孔道和纳米尺度的量子效应也引起了催化工作者的研究兴趣[14-15]。在多相催化体系中,载体的疏水性有利于吸附有机物,起到对底物富集的作用[16],载体的亲水性又可以使较为亲水的产物有效地从活性位点脱附,提高催化效率,而两亲性碳材料的制备及催化性能研究鲜有报道。
对氨基苯酚是一种重要的有机原料,广泛地应用于染料、医药、橡胶和感光材料等工业领域[17]。硝基苯催化加氢制备对氨基苯酚一般经历两个过程:首先硝基苯加氢生成中间体苯基羟胺(PHA),然后PHA在酸性介质中经Bamberger重排形成对氨基苯酚[18-19](图1)。与采用铁粉或硫化碱还原硝基苯相比,催化加氢法具有环境友好[16]、成本低、操作简便等优点[17-20],符合绿色化学原子经济性要求和环境保护要求。硝基苯经Bamberger重排是合成对氨基苯酚最便捷、经济的方法,是一种理想的生产工艺[21]。然而, PHA由于不易从活性位点脱附,易过度加氢生成大量的副产物苯胺[22-26]。因此,使PHA从活性位点脱附是提高对氨基苯酚选择性的有效措施。
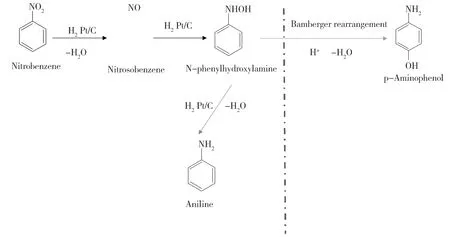
Fig.1 Process of Nitrobenzene hydrogenation图1 硝基苯加氢路线
本文采用Stöber法,以氟碳表面活性剂(FC4)和Pluronic F127在酸性乙醇溶液中形成的胶束作为模板,通过间苯二酚和甲醛共聚,经碳化,制备出具有明显的介孔孔道结构的介孔碳材料(C-x,x代表碳化温度),随后采用浸渍法吸附H2PtCl6,硼氢化钠作为还原剂,制备了催化剂Pt/C-400。该催化剂在硝基苯催化加氢制对氨基苯酚反应中,因其孔道和表面的两亲性有助于硝基苯催化加氢过程中间体从活性位点的脱附,提高了对氨基苯酚的选择性。
1 实验部分
1.1 试剂
负载量为5%的商业化铂碳(Pt/C)催化剂,硝基苯、间苯二酚(分析纯)购于上海阿拉丁生化科技股份有限公司;Pluronic F127(EO106PO70EO106)购于西格玛奥德里奇(中国)试剂公司;氯铂酸(H2PtCl6·6H2O)购于上海柏卡化学技术有限公司;氟碳表面活性剂(FC4)购于香港伊域化学。
1.2 实验方法
1.2.1 介孔碳材料制备
具有特殊介孔孔道结构碳材料的合成方法参见文献[7]。将1.0 g F127,0.2 g FC4和1.0 g KCl溶于60 mL 2.0 mol/L HCl水溶液中,在不断搅拌下加入1.0 g 1,3,5-三甲苯和15 mL乙醇,30 ℃下反应4 h。接着再加入0.4 g间苯二酚和0.56 mL 质量分数为35 %的甲醛水溶液,继续反应24 h后,将反应液转移至水热晶化釜中,100 ℃水热反应24 h。然后离心,将所得的固体材料用水和乙醇各洗3次,50 ℃真空干燥8 h,得到间苯二酚和甲醛酚醛树脂聚合物,记为RF。最后将所制备的RF进行碳化,在氮气保护下,先以5 ℃·min-1的升温速率升至200 ℃,维持2 h,再以1 ℃·min-1分别升至400 ℃、600 ℃,并维持3 h,分别得到介孔碳材料C-400、C-600。
1.2.2 Pt/C-400催化剂制备
将1.0 g H2PtCl6·6H2O溶于100 mL水中配制H2PtCl6水溶液(0.0193 mol/L)。取0.5 g C-400加入2.65 mL H2PtCl6水溶液和0.3 mL异丙醇溶液中进行浸渍,室温下搅拌至溶剂挥发,将所得材料于50℃真空条件下干燥 8 h,接着将上述材料加至5 mL异丙醇和0.5 mL乙醇的混合溶剂中,室温下加入适量NaBH4还原4 h。用蒸馏水和乙醇洗涤3~4次,50 ℃真空条件下干燥 8 h。得到催化剂Pt/C-400,Pt的负载量约为2%。
1.2.3 催化性能测试
硝基苯催化加氢制苯胺:将0.024 g Pt/C-400催化剂、2.5 mL甲苯和0.25 mL硝基苯加入至10 mL玻璃瓶,反应于高压反应釜中进行,用氢气置换三次后充入0.1 MPa H2,在 80℃反应4 h。反应结果用Agilent 7890A型气相色谱进行分析(毛细管柱:HP-5,30 m×0.32 mm×0.5 μm,FID检测器)。在相同的实验条件下,以5%的商业化Pt/C为催化剂进行上述反应的对照实验。
硝基苯催化加氢制对氨基苯酚:将0.024 g Pt/C-400催化剂、0.25 mL 硝基苯、2.5 mL甲苯和3.5 mL 质量分数为10% 的H2SO4加入10 mL玻璃瓶,反应于高压反应釜中进行,用氢气置换三次后充入0.1 MPa H2,在80 ℃反应4 h。反应结果用岛津高效液相色谱进行分析(色谱柱:C18柱,5 μm,4.6 mm×250 mm;柱温:30 ℃;紫外检测器:254 nm,V(CH3OH)∶V(H2O)=30∶70,进样量20 μL.)。以5%的商业化Pt/C为催化剂进行上述反应的对照实验。
1.2.4 表征条件
采用日本JEOL-JSM-6700F型扫描电子显微镜(SEM)和日本JEM-1011 100 kV透射电子显微镜(TEM)对样品的形貌和微观结构进行观察;采用美国ASAP2020全自动比表面及孔隙度分析仪进行样品的N2吸附-脱附测试,材料的比表面根据吸附支相对压力P/P0为0.05-0.15的数据按照Brunauer-Emmett-Teller (BET)方程计算,孔体积根据P/P0>0.99的数据进行计算。
2 结果与讨论
材料的制备过程如图2所示。第一步,F127和FC4表面活性剂在含有三甲苯的酸性乙醇水溶液中完成自组装,形成胶束;第二步,胶束和间苯二酚、甲醛缩合反应产物在乙醇的作用下通过氢键发生有机-有机自组装,初步形成有序介孔聚合物[7];第三步,通过100oC水热处理,完成进一步的酚醛聚合,形成具有独特结构的酚醛树脂聚合物(RF)。第四步,氮气氛围下,不同温度下碳化聚合物,得到介孔碳材料(C-x)。
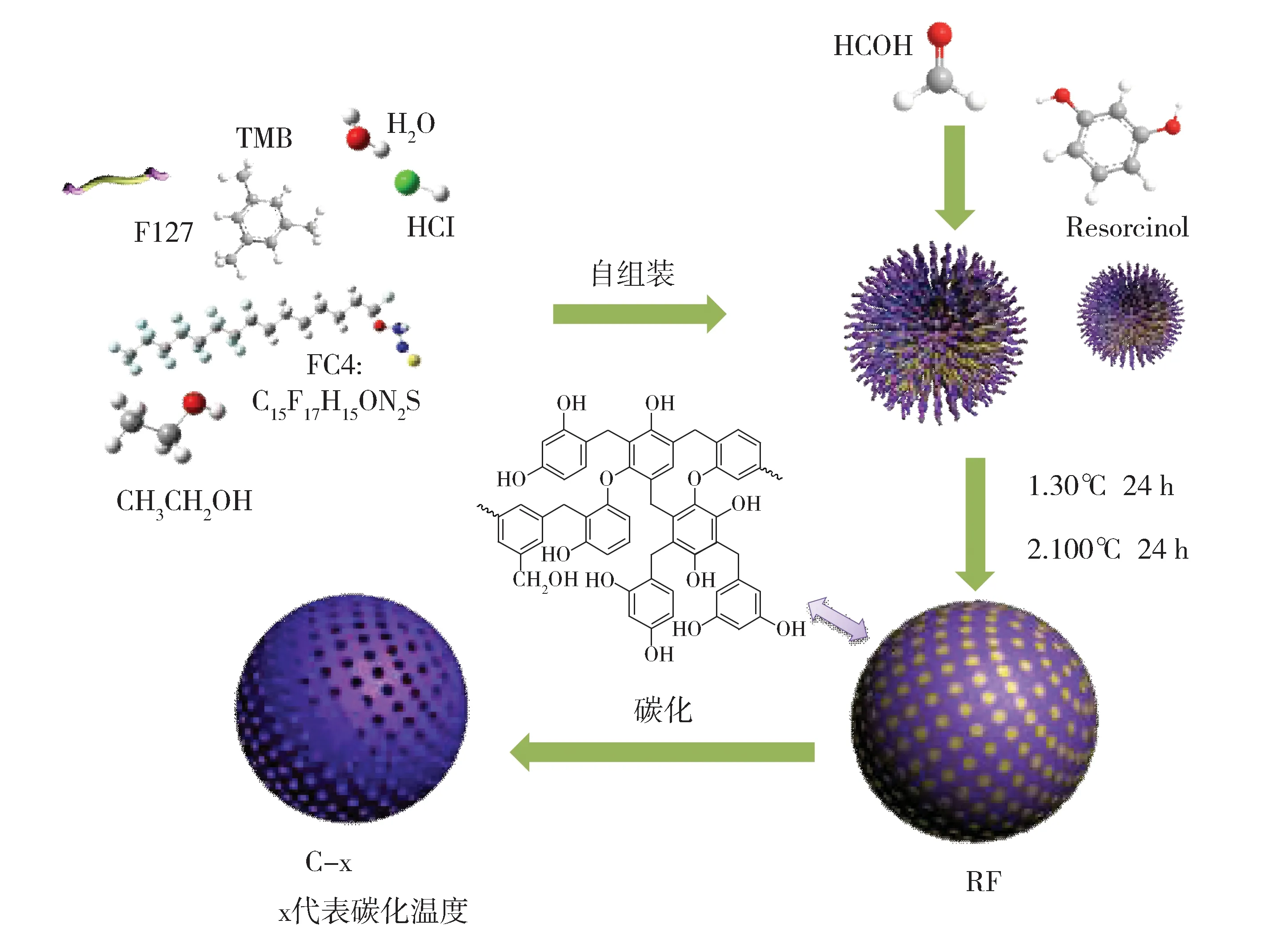
Fig.2 Schematic illustration of the formation process of C-x图2 C-x介孔碳球的制备示意图
2.1 结构表征
首先通过扫描电子显微镜(SEM)和透射电子显微镜(TEM)对RF进行微观形貌观察。从扫描电镜照片可以看出(图3a),RF为球形材料,直径为120~140 nm。图3b,3c为RF的TEM照片,从图片中可以清晰看到,RF结构中包含许多孔道。

Fig.3 Structure characterization of RF: (a) SEM image of RF; (b) and (c) TEM images of RF图3 RF的结构表征:(a) SEM照片;(b)和(c) TEM照片

Fig.4 Structure characterization of C-400 and C-600. (a) SEM image of C-400; (b) TEM images of C-400;(c) TEM image of C-600; (d) the nitrogen absorption desorption isotherms of C-400;(e) the pore-size distribution of C-400; (f) the nitrogen absorption desorption isotherms of C-600图4 C-400和C-600结构表征:(a) C-400的 SEM照片;(b) C-400的 TEM照片,插图:C-400局部放大;(c) C-600的TEM照片;(d) C-400的氮气吸附-脱附曲线;(e) C-400的孔径分布;(f) C-600的氮气吸附-脱附曲线
为了调控材料表面的亲疏水性,保留材料表面亲水基团,采用400 ℃、600 ℃分别进行碳化。采用SEM、TEM和N2吸附分别对C-400和C-600进行形貌和孔道结构表征(如图4所示)。图4a为C-400的SEM图,由图可知,C-400依然保持球形结构。图4b为其对应的TEM图片,图b插图为C-400的局部放大图,从图中可以看到,C-400与RF结构相似,粒径为120~140 nm,孔道排布更加清晰有序,孔道方向十分独特。这可能是因碳化过程引起骨架收缩造成的。通过N2吸附-脱附分析进一步对材料孔道进行表征,图4d为C-400的N2吸附-脱附曲线,从图中可以看出,C-400吸附曲线属于IV型,回滞环为H2型,说明材料具有典型的介孔孔道结构,图4e为其孔径分布图,孔径约为3.5 nm,BET 比表面积为 340.3 m2·g-1,孔体积为 0.29 cm3·g-1。图4c为C-600的TEM图,图中孔道骨架模糊,即RF在600℃碳化后,孔道结构很可能已经被破坏。为了进一步验证这一结果,对C-600进行N2吸附-脱附表征。 C-600的N2吸附-脱附曲线(如图4f)和BET比表面积(16.8 m2·g-1)进一步证明聚合物在600℃碳化时结构遭到破坏,孔道坍塌。综合介孔材料孔道对金属的限域作用和在传质过程中的优势,故选用C-400作为催化剂载体进行后续研究。
通过浸渍、NaBH4还原,制备了催化剂Pt/C-400,其结构表征结果如图5所示。从Pt/C-400的 TEM照片(图5a)可明显看出,大部分Pt纳米颗粒均匀地分散在载体的孔道内。Pt/C-400 N2吸附曲线为IV型(图5b),回滞环为H2型。Pt/C-400的BET比表面积为168.4 m2·g-1,孔体积为0.17 cm3·g-1。图5c为其孔径分布图,孔径约为4.0 nm,与C-400相近。与C-400相比,Pt/C-400除比表面积稍有下降外,负载金属后催化剂Pt/C-400基本保持了载体碳材料典型的介孔孔道结构,由于负载金属,造成了Pt/C-400比表面和孔体积下降。
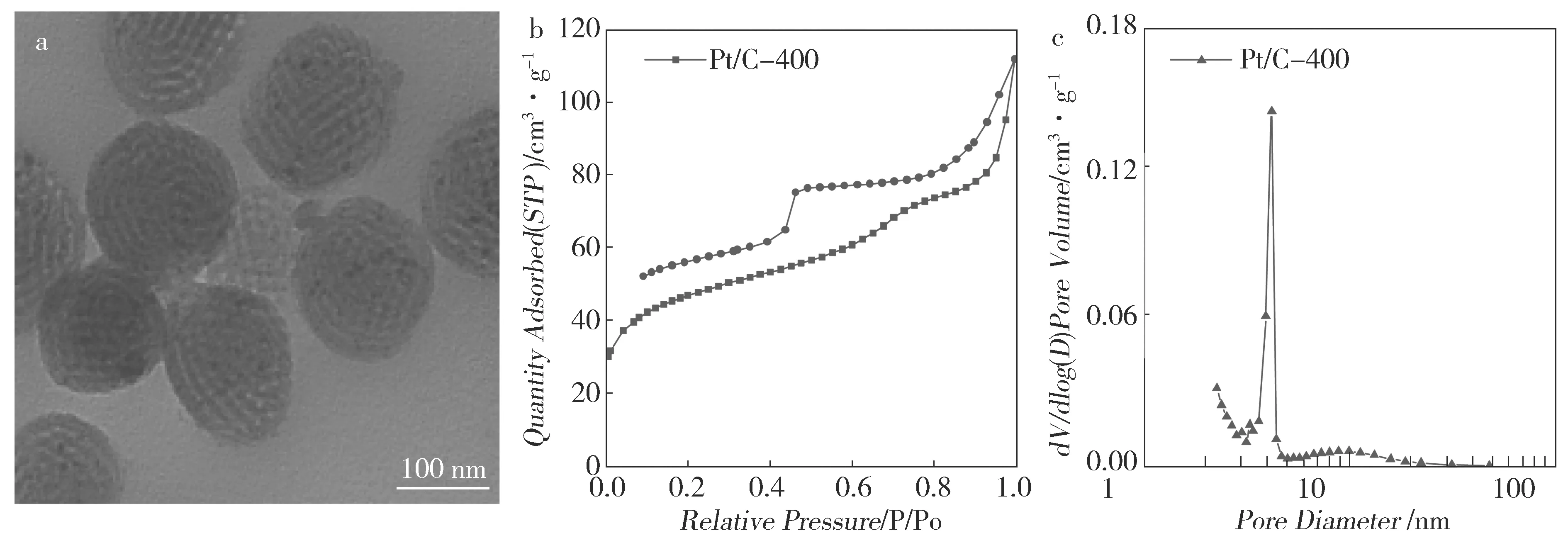
Fig.5 Structure characterization of Pt/C-400. (a) the TEM image of Pt/C-400; (b) the nitrogen absorption desorption isotherms of Pt/C-400; (c) the pore-size distribution of Pt/C-400图5 Pt/C-400结构表征:(a)TEM照片;(b)氮气吸附-脱附曲线;(c)Pt/C-400孔径分布
2.2 催化性能评价
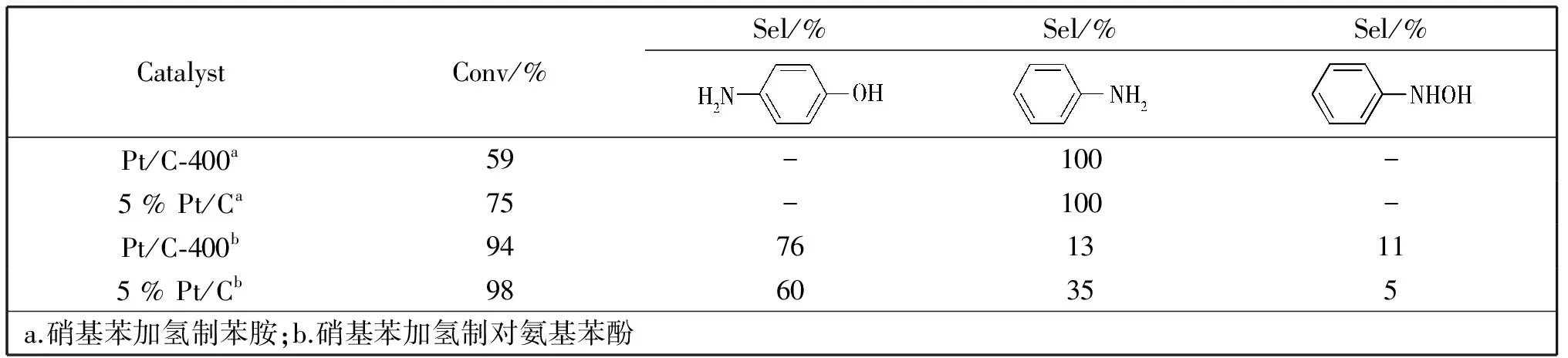
表1 不同催化剂催化性能比较Table 1 Comparison of catalytic performance of different catalysts
以硝基苯加氢制苯胺和硝基苯加氢制对氨基苯酚为模型反应,对Pt/C-400的催化活性和选择性进行了评价。反应按照n(硝基苯)∶n(Pt)=1 000,80℃,0.1 MPa H2条件下反应4 h。实验结果如表1所示。
在硝基苯催化加氢制苯胺的反应中,以Pt/C-400为催化剂时,硝基苯转化率为59%,相同条件下以5%的商业化Pt/C为催化剂时,硝基苯的转化率为75%,Pt/C-400在保证有适当转化率的前提下减缓了加氢速率,有助于控制加氢反应和重排反应的相对速率。
在硝基苯催化加氢到对氨基苯酚的实验中,以Pt/C-400为催化剂的体系,硝基苯转化率为94%时对氨基苯酚选择性可达76%,中间体PHA的选择性为11%,在相同反应条件下,以5%的商业化Pt/C为催化剂时,硝基苯的转化率可达98%,但对氨基苯酚的选择性仅有60%,中间体的选择性为5%,与5% 的Pt/C 催化剂相比,虽然硝基苯的转化率稍低,但是对氨基苯酚的选择性明显升高,且由中间体PHA的含量可以看出,以Pt/C-400为催化剂的体系中PHA更易从催化剂脱附,向体相扩散,有利于对氨基苯酚的形成。Pt/C-400催化剂的高活性和高选择性极有可能与其催化剂的孔道结构和载体特殊的表面性质有关。Pt/C-400具有孔分布集中、孔道排列有序的介孔孔道,这种介孔的孔道结构一方面可以增加材料的比表面积,提高金属在载体上的分散性,而且载体与金属间相互作用有利于增强金属颗粒在载体上的稳定性;另一方面,Pt/C-400的孔径较大(4.0 nm),有利于底物的扩散,减小了传质阻力,同时也有利于PHA从金属表面脱附向体相扩散,有利于参与Bamberger重排[16]形成对氨基苯酚,避免PHA进一步加氢生成苯胺,进而提高了选择性。

Fig.6 Amphipathic test of (a)RF;(b) Pt/C-400;(c) 5% Pt/C图6 亲疏水性测试:(a)RF;(b) Pt/C-400;(c) 5% Pt/C
为了验证催化剂表面性质的差异,采用甲苯-水体系对材料表面的亲疏水性进行了测试,如图6。在10 mL小瓶中加入1.5 mL H2O,然后分别加入0.02 g RF、Pt/C-400和5%的商业化Pt/C催化剂,再加入1.5 mL 甲苯,轻轻晃动小瓶,观察材料在两相的分散情况。图6a为RF在水/甲苯两相体系中的分散情况照片,可以看到,RF完全分散在下层的水相中,表明其有较强的亲水性,这是由于在碳化前聚合物表面含有大量的羟基使得材料表面具有亲水性;图6b为Pt/C-400在水/甲苯两相体系中的分散情况照片,从中可以清晰看出Pt/C-400分布于油水两相界面,说明材料具有两亲性,这可能是因为400℃碳化过程中没有完全烧掉材料中的—OH,保留的—OH使得Pt/C-400具有表面两亲性。如图6c所示,5%的商业化Pt/C催化剂则几乎完全分散在有机相中,说明5%的商业化Pt/C表面疏水。结合催化活性评价和亲疏水性实验结果,表明Pt/C-400催化剂的表面两亲性一方面有利于硝基苯的吸附,能有效降低反应的传质阻力,保证了硝基苯一定的加氢速率,另一方面有利于中间体PHA从活性位点脱附,避免了PHA深度加氢到苯胺的副反应,提高了反应的选择性。
3 结论
以表面活性剂FC4和F127自组装形成的胶束为模板,经400 ℃碳化,负载金属Pt,成功制备了Pt/C-400催化剂。在硝基苯制对氨基苯酚反应中,与负载量为5%的商业化Pt/C催化剂相比,Pt/C-400表现出较好的催化性能,硝基苯的转化率和对氨基苯酚的选择性可分别达到94%和76%。其原因一方面是Pt/C-400具有介孔孔道结构,使得金属在载体上很好地分散,减小底物向活性位点扩散和中间体脱附催化剂向体相扩散的传质阻力。另一方面是催化剂的表面两亲性既有利于催化剂对底物的吸附,又有利于中间体从活性位点的脱附,避免PHA深度加氢,有利于Bamberger 重排过程,从而提高了转化率。
[1] Yan Y,Zhang F Q,Meng Y,etal.One-step Synthesis of Ordered Mesoporous Carbonaceous Spheres by an Aerosol-assisted Self-assembly[J].ChemCommun,2007:2867-2869.DOI:10.1039/b702232h.
[2] Liu J,Wickramaratne N P,Qiao S Z,etal.Molecular-based Design and Emerging Applications of Nanoporous Carbon Spheres[J].NatMter,2015,14(8):763-774.DOI:10.1038/nmat4317.
[3] Liang C D,Li Z J,Dai S.Mesoporous Carbon Materials:Synthesis and Modification[J].AngewChemInEd,2008,47(20):3696-3717.DOI:10.1002/anie.200702046.
[4] Wan Y,Shi Y F,Zhao D Y.Supramolecular Aggregates as Templates:Ordered Mesoporous Polymers and Carbons[J].ChemMater,2008,20:932-945.DOI:10.1021/cm7024125.
[5] Tanaka S,Nishiyama N,Egashira Y,etal.Synthesis of Ordered Mesoporous Carbons with Channel Structure from an Organic-organic Nanocomposite[J].ChemCommun,2005:2125-2127.DOI:10.1039/b501259g.
[6] Zhang F Q,Meng Y,Gu D,Yan Y,etal.A Facile Aqueous Route to Synthesize Highly Ordered Mesoporous Polymers and Carbon Frameworks with Ia3d Bicontinuous Cubic Structure[J].JAmChemSoc,2005,127:13508:13509.DOI:10.1021/ja0545721.
[7] Liu J,Qiao S Z,Liu H,etal.Extension of the Stober Method to the Preparation of Monodisperse Resorcinol-formaldehyde Resin Polymer and Carbon Spheres[J].AngewChemInEd,2011,50(26):5947-5951.DOI:10.1002/anie.201102011.
[8] Liu J,Yang T Y,Wang D W,etal.A Facile Soft-template Synthesis of Mesoporous Polymeric and Carbonaceous Nanospheres[J].NatCommun,2013,4.DOI:10.1038/ncomms3798.
[9] Liu R L,Shi Y F,Wan Y,etal.Triconstituent Co-assembly to Ordered Mesostructured Polymer-Silica and Carbon-Silica Nanocomposites and Large-Pore Mesoporous Carbons with High Surface Areas[J].JAmChemSoc,2006,128:11652-11662.DOI:10.1021/ja0633518.
[10] Yang H F,Shi Q H,Liu X Y,etal.Synthesis of Ordered Mesoporous Carbon Monoliths with Bicontinuous Cubic Pore Structure of Ia3d Symmetry[J].ChemCommun,2002:2842-2843.DOI:10.1039/b209233f.
[11] Liu J Y,Song Y H,Xu H,etal.Non-metal Photocatalyst Nitrogen-doped Carbon Nanotubes Modified Mpg-C3N4:Facile Synthesis and the Enhanced Visible-light Photocatalytic Activity[J].JournalofColloidandInterfaceScience,2017,494:38-46.DOI:10.1016/j.jcis.2017.01.010.
[12] Wang G H,Cao Z,Gu D,etal.Nitrogen-Doped Ordered Mesoporous Carbon Supported Bimetallic PtCo Nanoparticles for Upgrading of Biophenolics[J].AngewChemIntEd,2016,55(31):8850-8855.DOI:10.1002/anie.201511558.
[13] 闫菊平,王英特,陈莉,等.碳点的制备及其光电响应的研究[J].山西大学学报:自然科学版,2016,39(151):108-112.DOI:10.13451/j.cnki.shanxi.univ(nat.sci.).2016.01.018.
[14] Liu H.Size Effect of Quantum Conductance in Carbon Nanotube Y-Junctions[J].ChinPhysB,2010,10:057206.
[15] Chen Binhao,etal.Quantum Effects on Adsorption Isotherm of Hydrogen in Strongly Confining Twisted Carbon Nanotubes[J].InterJHydrogenEnergy,2015,40(38):12993-3002.DOI:10.1016/j.ijhydene.2015.07.144.
[16] Yang H Q,Jiao X,Li S R,etal.Hydrophobic Core-hydrophilic Shell-structured Catalysts:A General Strategy for Improving the Reaction Rate in Water[J].ChemCommun,2012,48:11217-11219.DOI:10.1039/c2cc36273b.
[17] 王莲鸳,程振兴,薛勇,等.硝基苯加氢合成对氨基酚用负载铂催化剂的制备[J].催化学报,2001,23:19-23.
[18] Min K I,Choi J S,Chung Y M,etal.p-Aminophenol Synthesis in an Organic/Aqueous System using Pt Supported on Mesoporous Carbons[J].AppliedCatalysisA:General,2008,337(1):97-104.DOI:10.1016/j.apcata.2007.12.004.
[19] Nadgeri J M,Biradar N S,Patil P B,etal.Control of Competing Hydrogenation of Phenylhydroxylamine to Aniline in a Single-Step Hydrogenation of Nitrobenzene top-Aminophenol[J].IndEngChemRes,2011,50(9):5478-5484.DOI:10.1021/ie102544a.
[20] Rode C V,Vaidya M J,Chaudhari R V,etal.Synthesis of Aminophenol by Catalytic Hydrogenation of Nitrobenzene[J].OrgProcessResDe,1999,3:465-470.DOI:10.1021/op990040r.
[21] 徐梅君.硝基苯加氢还原制对氨基苯酚的工艺探讨[J].辽宁化工,1993:37-40.
[22] 刘东志,陈昌藻.Pt-活性炭催化剂催化硝基苯加氢制备对氨基酚[J].催化学报,1996,17(6):544-546.
[23] Zhang T T,Jiang J Y,Wang Y H.Green Route for the Preparation ofp-Aminophenol from Nitrobenzene by Catalytic Hydrogenation in Pressurized CO2/H2O System[J].OrgProcessResDe,2015,19(12):2050-2054.DOI:10.1021/acs.oprd.5b00307.
[24] Komatsu Takayuki,Hirose Tatsuo.Gas Phase Synthesis of Para-aminophenol from Nitrobenzene on Pt/zeolite Catalysts[J].AppliedCatalysisA:General,2004,276:95-102.DOI:10.1016/j.apcata.2004.07.044.
[25] Quartarone G,Ronchin L,Tosetto A,etal.New Insight on the Mechanism of the Catalytic Hydrogenation of Nitrobenzene to 4-aminophenol in CH3CN—H2O—CF3COOH as a Reusable Solvent System.Hydrogenation of Nitrobenzene Catalyzed by Precious Metals Supported on Carbon[J].AppliedCatalysisA:General,2014,475:169-178.DOI:10.1016/j.apcata.2014.01.033.
[26] Liu P L,Hu Y H,Ni M,etal.Liquid Phase Hydrogenation of Nitrobenzene to Para-Aminophenol over Pt/ZrO2Catalyst and SO42-/ZrO2-Al2O3Solid Acid[J].CatalLett,2010,140:65-68.DOI:10.1007/s10562-010-0427-8.
AmphiphilicMesoporousCarbonMaterialloadedPtasaCatalysttowardtheTransformationofNitrobenzenetoP-aminophenol
LIANG Jifen1,HAO Yajuan2,YANG Hengquan2*
(1.InstituteofMolecularScience,ShanxiUniversity,Taiyuan,Shanxi030006,China;2.SchoolofChemistryandChemicalEngineering,ShanxiUniversity,Taiyuan030006,China)
Mesoporous carbon nanospheres were prepared successfully using fluorocarbon surfactant (FC4) and Pluronic F127 as templates in acidic ethanol solution followed by carbonization under N2atmosphere. Pt nanoparticles were introduced via a traditional impregnation method. The structure of synthesized material and catalyst were fully characterized by scanning electron microscope (SEM), transmission electron microscope (TEM), N2adsorption-desorption, and their amphiphilic properties were tested through oil-water distribution experiment. The results showed that an amphiphilic catalyst Pt/C-400 with uniform Pt particle size distribution and unique pore structure could be got by controlling the carbonization temperature. Using Pt/C-400 as catalyst, nitrobenzene could be converted to p-aminophenol with 94 % of conversion and 76 % of selectivity. Compared with the commercial 5 % (metal loadings fraction) Pt/C catalyst the content of intermediate product and the selectivity of N-phenylhydroxlamine were much higher, which might be attributed to the unique pore structure and amphipathic interfaces.
amphipathic mesoporous carbon;hydrogenation of nitrobenzene;bamberger rearrangement;fluorocarbon surfactant
10.13451/j.cnki.shanxi.univ(nat.sci.).2017.04.027
2017-03-20;
2017-04-14
国家自然科学基金(201573136;U1510105);山西大学科研启动基金(rsc723)
梁继芬(1990-),女,山西太原人,硕士研究生,主要从事催化材料的制备和性能方面的研究。E-mail:18734150091@163.com
*通信作者:杨恒权(YANG Hengquan),E-mail:hqyang@sxu.edu.cn
O643.3
A
0253-2395(2017)04-0846-08
猜你喜欢
云南化工(2022年9期)2022-10-12
武汉纺织大学学报(2021年6期)2022-01-05
中国生物医学工程学报(2019年4期)2019-07-16
环境科技(2017年6期)2018-01-17
当代化工研究(2016年2期)2016-03-20
合成化学(2015年2期)2016-01-17
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
应用化工(2014年1期)2014-08-16
应用化工(2014年4期)2014-08-16
无机化学学报(2014年4期)2014-02-28
