
单纯性甲基丙二酸血症一例及家系MUT基因突变分析
2018-01-02 07:50梁如佳魏彦敏
临床误诊误治 2017年12期
梁如佳,魏彦敏,刘 芳
单纯性甲基丙二酸血症一例及家系MUT基因突变分析
梁如佳,魏彦敏,刘 芳
目的探讨甲基丙二酸血症(methylmalonic acidemia, MMA)家系MUT基因的临床特点。方法对诊治的1例单纯性MMA的临床资料及家系MUT基因突变情况进行回顾性分析。结果男,52 d。系第3胎第3产,其母孕期体健,父母非近亲结婚,第1胎为足月顺产女婴,出生7 d后去世,当时考虑酸中毒,未行遗传代谢病检查;第2胎亦为足月顺产女婴,体健。本例于生后36 d出现抽搐,症状持续10 min缓解,就诊当地医院,入院时呈昏迷状态,伴严重代谢性酸中毒,请我院新生儿科会诊,高度怀疑遗传代谢疾病,急诊采血留尿行遗传代谢病筛查,同时予维生素B12、左卡尼汀等治疗。遗传代谢病筛查结果支持MMA诊断,遂继续上述治疗,后病情平稳出院。出院后患儿父母擅自停药并停用特殊奶粉,20 d后再次昏迷,家属放弃治疗,数日后死亡。通过对患儿及其父母、姐姐进行相关基因高通量测序,发现患儿父亲MUT基因发生了错义突变(c.1106G>A),母亲发生了杂合突变(c.729_730insTT)。结论基因测序有助于明确MMA的临床分型,为产前诊断奠定基础,避免缺陷患儿出生。
甲基丙二酸血症;甲基丙二酸单酰CoA变位酶;突变
甲基丙二酸血症(methylmalonic acidemia, MMA)又称甲基丙二酸血尿症,由Oberholzer等[1]率先发现并报道,为常染色体隐性遗传病,是新生儿先天性有机酸代谢异常疾病之一,发病率为1∶250000~1∶48000[2]。随着分子生物学技术的进展,尤其是基因测序技术的发展,使MMA确诊及分型更为精准,同时为产前诊断奠定了基础。我院近期收治1例MMA,现分析报告如下。
1 临床资料
1.1病例资料 男,52 d。系第3胎第3产,孕40周顺产娩出,出生体重3 kg,无胎膜早破,羊水、胎盘、脐带无异常,阿氏评分不详,否认窒息抢救史。其母孕期体健,无高血压病、糖尿病,否认肝炎、结核等传染病病史。父母非近亲结婚,第1胎为足月顺产女婴,因疑诊酸中毒于出生7 d后去世,未行遗传代谢病检查;第2胎为足月顺产女婴,体健。本例出生不久因呕吐、吃奶欠佳就诊当地医院,按新生儿咽下综合征、新生儿肺炎、心肌损害、新生儿高胆红素血症、低钾血症等住院治疗7 d后痊愈出院。出院后家属自行看护,混合喂养。出生后36 d患儿出现抽搐,症状持续10 min缓解,立即就诊当地医院,入院时呈昏迷状态,伴严重代谢性酸中毒,请我院新生儿科会诊,高度怀疑遗传代谢疾病,急诊采血留尿行遗传代谢病筛查,同时予维生素B121 mg/d肌内注射,静脉滴注左卡尼汀200 mg/d,同时予纠正代谢性酸中毒等治疗。5 d后遗传代谢病筛查结果支持MMA诊断,继续原治疗方案,患儿病情渐好转,予特殊奶粉混合喂养,因家长强烈要求,于治疗10 d后出院。出院后家长自行停用所有药物,且停止特殊奶粉喂养。20 d后患儿再次昏迷,到当地卫生院就诊,因病情重,家属放弃治疗,数日后死亡。
1.2基因检测 经患儿家属同意后进行MMA基因检测,采集患儿及其父母、姐姐静脉血2 ml,置于乙二酸四己盐酸(EDTA)抗凝剂的采血管中,-20℃保存待检。选用新一代测序仪Illumina NextSeq 500进行高通量测序,同时对DNA样本进行相关基因及生物信息分析,并与SNP、InDel等公共数据库进行对比,以获得可疑位点,确定可疑来源。
2 结果
2.1遗传代谢疾病检测 利用气相色谱/质谱串联技术(GC/MS)检测患儿血丙酰肉碱52.22 mol/L(正常参考值0.3~5.0 mol/L),丙酰肉碱/乙酰肉碱 3.50(正常参考值0.02~0.25)。尿气相色谱-质谱检测尿甲基丙二酸为55.6 g/mol(正常参考值0~4.0 g/mol),甲基枸橼酸22.3 g/mol(正常参考值0~0.7 g/mol),确诊为MMA。
2.2基因测序结果 本例MUT基因有2个杂合突变:①c.729_730insTT(插入胸腺嘧啶),导致氨基酸改变p.D244Lfs*39(移码突变)。患儿母亲该位点为杂合变异,父亲则无变异。②c.1106G>A(编码区第1106号核苷酸由鸟嘌呤变异为腺嘌呤),致第369号氨基酸由精氨酸变异为组氨酸,即p.R369H(错义突变),其不属于多态性位点,发生率极低。患儿母亲该位点无变异,父亲则为杂合变异。患儿及其父母、姐姐MUT基因测序见图1~4。
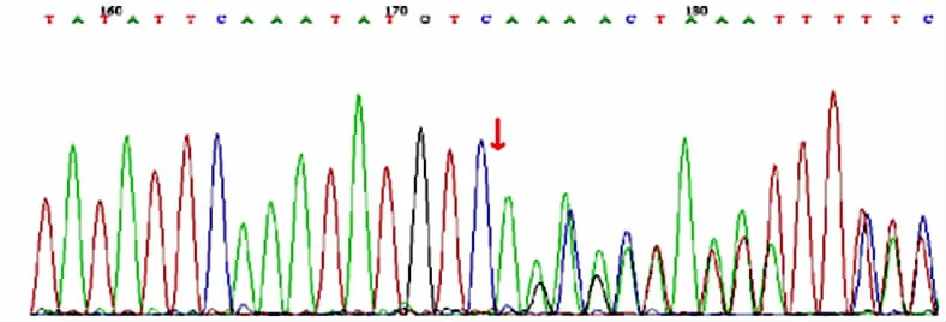
图1单纯甲基丙二酸血症患儿母亲突变基因DNA测序图

图2单纯甲基丙二酸血症患儿父亲突变基因DNA测序图
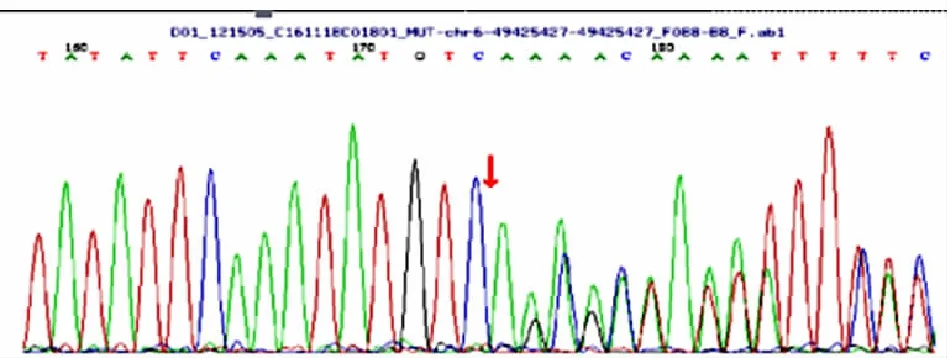
图3单纯甲基丙二酸血症患儿突变基因DNA测序图
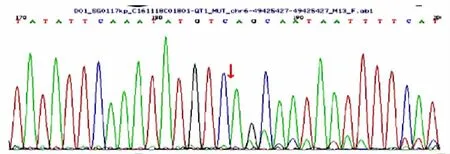
图4单纯甲基丙二酸血症患儿姐姐突变基因DNA测序图
3 讨论
3.1疾病概述 MMA属于常染色体隐性遗传病,是我国新生儿期最常见的有机酸血症,不同国家和地区的发病率不同。有研究显示,美国MMA发病率为1/2.9万,加拿大为1/1.6万,台湾为1/8.5万,北京为1/2.6万,浙江为1/6.4万[3-4]。韩连书等[5]通过串联质谱技术对国内MMA患儿进行筛查,发现国内MMA发病率为1/5.7534万。
MMA是体内支链氨基酸、胆固醇和奇数链脂肪酸在分解代谢过程中因甲基丙二酰辅酶变位酶(MCM)或其辅酶钴胺素(Ado-Cbl,维生素B12)代谢功能障碍,致琥珀酸生成通路受阻,旁路代谢增强,造成毒性物质(甲基丙二酸、3-羟基丙酸、丙二酸及甲基枸橼酸等)异常累积,引起肝、肾、神经等损伤[6]。
3.2临床分型 根据病因将MMA分为MCM代谢缺陷和维生素B12代谢缺陷两大类,其中前者分为完全缺陷(mut0)型和部分缺陷(mut-)型,后者分为线粒体钴胺素还原酶(cb1A)缺乏、钴胺素腺苷转移酶(cb1B)缺乏及由胞质和溶酶体钴胺素代谢异常引起的腺苷钴胺素和甲基钴胺素合成缺陷(cb1C、cb1D、cb1F)型[7]。mut0、mut-、cblA、cblB型MMA患儿临床仅表现为血MMA水平升高,称为单纯性MMA;cb1C、cb1D、cb1F型MMA患儿除血MMA水平升高,同时合并血同型半胱氨酸水平升高,称为MMA并同型半胱氨酸血症(合并型)。国内文献报道,约30%的患儿表现为单纯性MMA,70%为合并型MMA[8]。根据患儿对维生素B12治疗的有效性,临床可将MMA分为维生素B12治疗有效型和无效型[9]。mut0、mut-为维生素B12治疗无效型,cblA、cblB对维生素B12治疗小部分有效,cb1C、cb1D、cb1F对维生素B12治疗大部分有效。
3.3基因分型及临床表现 目前已发现4种基因编码的酶缺陷可导致单纯型MMA,分别为MUT、MMAA、MMAB和MMADHC基因,其中MUT基因突变是国内外最常见的突变类型[10]。MCM由MUT基因编码[11]。文献报道MUT基因突变类型有250多种[12],已证实单纯型MMA多由MUT基因突变引起[13]。MUT基因突变可引起MCM活性降低,导致患儿出现一系列临床症状。由MUT基因突变引起的MMA,患儿多对维生素B12治疗无效,且80%患儿在出生1周内出现症状,90%患儿于出生1个月内出现症状,病死率及并发症发生率极高[14]。有报道发现mut0型MMA患儿对维生素B12治疗无效,预后最差,其中60%的患儿死亡[15]。mut-型MMA患儿对维生素B12的治疗效果优于mut0型,且多数患儿于出生后1个月出现临床症状[5,16]。MMA患儿发病年龄不尽相同,从新生儿期到成人期均可发病,且发病年龄越小,病情越危重,预后越差。婴儿期发病的MMA患儿常见临床表现为吃奶欠佳、反应差、嗜睡、肌张力降低、发育迟缓等,实验室检查则表现为难以纠正的代谢性酸中毒、高氨血症、中性粒细胞减少等[17]。
3.4诊断 MMA诊断应结合下列情况:①一般实验室检测:血常规、尿酮测定、血气分析、血氨、乳酸等检查;②串联质谱检测:测定血丙酰基肉碱、丙酰基肉毒碱/乙酰基肉毒碱、游离肉碱、丙酰基肉毒碱/游离肉碱、蛋氨酸水平;③有机酸分析:测定尿甲基丙二酸、甲基枸橼酸水平;④检测血清及尿液总同型半胱氨酸水平;⑤维生素B12负荷试验:检测患儿是否对维生素B12治疗有效;⑥酶学分析:检测外周血淋巴细胞、皮肤成纤维细胞中MCM的活性;⑦基因检测:先证者的基因检测可明确临床分型,为治疗提供最佳方案[18]。
3.5治疗 临床治疗MMA以减少代谢产物累积,加快清除速率为基本原则。急性发作期MMA患儿以纠酸补液、低蛋白高热量饮食为主,同时限制蛋白质摄入,保障营养支持。对维生素B12治疗有效型患儿长期坚持维生素B12肌内注射,同时每日补充左旋肉碱,一方面促进体内甲基丙二酸、游离肉碱排出体外,一方面增强机体对蛋白质的抵抗力。合并型MMA必要时可补充叶酸。MMA患儿饮食以低蛋白、高热量为主,喂养特殊配方奶粉(即不含亮氨酸、异亮氨酸、缬氨酸、苏氨酸和蛋氨酸的奶粉),避免甲基丙二酸前体氨基酸的摄入。
3.6基因检测的临床意义 新生儿期遗传代谢病筛查是婴儿健康成长的一个重要保障,且MMA是可早发现、早诊断、早干预、早治疗的遗传代谢病之一。因MMA为常染色体隐性遗传病,若该患儿父母再次生育,分娩出MMA患儿的概率为1/4,在明确患儿诊断及父母基因突变情况的基础上,进行产前诊断尤为重要。目前临床大部分MMA患儿未进行基因检测,无法明确分型,对下一胎的产前诊断十分不利。随着新生儿筛查技术的普及,MMA检出率明显提高,但仅通过串联质谱检查和尿液有机酸分析已不能确定临床分型,尤其是部分致死型患儿,基因突变分析是确定分型最可靠的依据,为临床提供最佳治疗方案。此外,指导MMA家系进行产前诊断,避免下一胎出生缺陷,实现优生优育。本例予维生素B12治疗后症状明显减轻,证明该患儿为mut-型MMA,且基因分析亦显示为mut-型。
综上,基因测序可在明确临床分型的基础上采取针对性治疗措施,提高疗效,同时对再生育胎儿进行相对基因位点测序,明确是否有基因突变,对预防出生缺陷儿意义重大。
[1] Oberholzer V G, Levin B, Burgees E A,etal. Methylmalonic aciduria. An inborn error of metabolism leading to chronic metabolic acidosis[J].Arch Dis Child, 1967,42(225):492-504.
[2] Wang F, Han L, Yang Y,etal. Clinical, biochemical, and molecular analysis of combined methymalonic acidemia and hyperhomocysteinemia (cblC type) in China[J].J Inherit Metab Dis, 2010,33(S3):435-442.
[3] Ma X, Zhang Y, Yang Y,etal. Epilepsy in children with methylmalonic acidemia: electroclinical features and prognosis[J].Brain Dev, 2011,33(9):790-795.
[4] Han B, Cao Z, Tian L,etal. Clinical presentation, gene analysis and outcomes in young patients with early-treated combined methylmalonic acidemia and homocysteinemia (cblC type) in Shandong province, China[J].Brain Dev, 2016,38(5):491-497.
[5] 韩连书,毋盛楠,叶军,等.单纯型甲基丙二酸血症患者诊治分析[J].中华医学遗传学杂志,2013,30(5):589-593.
[6] 李璃,马定远,孙云,等.应用Ion Torrent测序技术检测一个甲基丙二酸血症家系的致病突变暨产前诊断[J].中华医学遗传学杂志,2016,33(2):181-185.
[7] 黎芳,麻宏伟,宋莹,等.甲基丙二酸血症的临床特点及基因型与临床表型关系的探讨[J].中国儿童保健杂志,2013,21(4):348-350.
[8] 刘玉鹏,王海军,吴桐菲,等.甲基丙二酸尿症cblB型1例及其MMAB基因新突变[J].中国当代儿科杂志,2015,17(2):172-175.
[9] Deodato F, Boenzi S, Santorelli F M,etal. Methylmalonic and propionic aciduria[J].AM J Med Genet C Semin Med Genet, 2006,142(2):104-112.
[10] Liu M Y, Liu T T, Yang Y L,etal. Mutation profile of the MUT gene in Chinese methylmalonic aciduria patients[J].JIMD Rep, 2012,6:55-64.
[11] 孙云,杨冰,焦泽霖,等.串联质谱技术新生儿筛查发现甲基丙二酸血症1例并文献复习[J].南京医科大学学报(自然科学版),2014,34(10):1451-1453.
[12] Forny P, Froese D S, Suormala T,etal. Functional characterization and categorization of missense mutations that cause methylmalonyl-CoA mutase (MUT) deficiency[J].Hum Mutat, 2014,35(12):1449-1458.
[13] 蔡奥捷,宗亚楠,刘宁,等.单纯型甲基丙二酸血症家系MUT基因分析及在产前诊断中的应用[J].中华围产医学杂志,2016,19(9):688-694.
[14] 郑雷,童凡.影响甲基丙二酸血症患儿临床疗效及预后的分子生物学机制分析[J].中国儿童保健杂志,2015,23(11):1169-1171.
[15] Cosson M A, Benoist J F, Touati G,etal. Long-term outcome in methylmalonic aciduria: a series of 30 French patients[J].Mol Genet Metab, 2009,97(3):172-178.
[16] Froese D S, Gravel R A. Genetic disorders of vitamin B12metabolism: eight complementation groups-eight genes[J].Expert Rev Mol Med, 2010,12:37.
[17] 赵正言.顾学范.新生儿遗传代谢病筛查[M].北京:人民卫生出版社,2015:185-188.
[18] 胡亚美,江载芳,申昆玲,等.诸福棠实用儿科学[M].北京:人民卫生出版社,2015:2284-2286.
AnalysisofOnePatientwithSimpleMethylmalonicAcidemiaandParentageMUTGeneMutation
LIANG Ru-jia1, WEI Yan-min2, LIU Fang3
(1. Chengde Medical College, Chengde, Hebei 067000, China; 2. Department of Neonates, Shenglujia Obstetric Hospital, Shijiazhuang 050000, China; 3. Department of Neonates, Bethune International Peace Hospital of PLA, Shijiazhuang 050082, China)
ObjectiveTo investigate clinical characteristics of methylmalonic acidemia (MMA) parentage MUT gene.MethodsClinical data of 1 patient with simple MMA and condition of parentage MUT gene mutation was retrospectively analyzed.ResultsA male neonate with 52 d old was the third child by the third births, and his parents were not consanguineous marriage. The first child was a full-term delivery girl, and she died the 7thd after birth, acidosis was suspected at that time without tests for genetic and metabolic diseases. The second child was also a full-term delivery girl with healthy. The third child had convulsion at the postnatal 36thd, and the symptom was relieved after continue 10 minutes. He visited in local hospital, and he was in comatose state when he had been admitted with severe metabolic acidosis, and the consultation with our hospital was required. Genetic metabolic disease was highly suspected, and screening for genetic metabolic diseases was performed by drawing blood and remaining urine in emergency department, and treatments such as vitamin B12 and Levocarnitine were given simultaneously. Screening result for genetic metabolic diseases supported the diagnosis of MMA, and the above treatments were continued, and then the neonate was discharged after having stable condition. After discharging, the parents stopped using the drug and special milk powder without doctors's authorization, and the neonate had coma again at the 20thd after discharging, and the family members gave up treatment, and then the neonate died a few days later. Related gene high-throughput sequencing was performed for the neonate and the neonatal parents and sister, and then the missense mutation (c.1106G>A) was detected in the neonatal father; heterozygosis mutation (c.729_730insTT) was detected in the neonatal mother.ConclusionGene sequencing helps to confirm clinical MMA classification, and it gives prepare for prenatal diagnosis to avoid defects in children born.
Methylmalonic acidemia; Methylmalonyl-CoA mutase; Mutation
067000 河北 承德,承德医学院(梁如佳);050000 石家庄,石家庄圣禄嘉妇产医院新生儿科(魏彦敏);050082 石家庄,解放军白求恩国际和平医院新生儿科(刘芳)
刘芳,E-mail:liufanglafy@126.com
R596.1
A
1002-3429(2017)12-0037-04
10.3969/j.issn.1002-3429.2017.12.016
2017-08-02 修回时间:2017-09-14)
猜你喜欢
有机氟工业(2022年1期)2022-09-13
食品工业科技(2021年9期)2021-06-25
中国生殖健康(2020年2期)2021-01-18
探索科学(学术版)(2020年4期)2021-01-18
中国畜牧杂志(2019年4期)2019-04-20
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
家庭百事通·健康一点通(2017年3期)2017-03-22
灾害医学与救援(电子版)(2016年2期)2016-03-11
医学研究杂志(2015年3期)2015-06-10
