
脊肌萎缩症合并I型呼吸衰竭1例临床及家系基因分析☆
2017-12-14 03:15张丽丹徐玲玲梁玉坚黄雪琼程玉才裴瑜馨张成唐雯
中国神经精神疾病杂志 2017年9期
张丽丹 徐玲玲 梁玉坚 黄雪琼程玉才 裴瑜馨 张成 唐雯○☆
脊肌萎缩症合并I型呼吸衰竭1例临床及家系基因分析☆
张丽丹*徐玲玲*梁玉坚*黄雪琼*程玉才*裴瑜馨*张成**唐雯*○☆
脊髓性肌萎缩症合并呼吸衰竭1型 家系分析IGHMBP2基因
脊肌萎缩症合并I型呼吸衰竭 (spinal muscular atrophy with respiratory distress type 1,SMARD1)是一种常染色体隐性遗传性疾病,该病非常罕见,至今全世界已报告60余例,而我国仅有1例报告。致病基因位于常染色体11q13.3的免疫球蛋白μ结合蛋白2 (immunoglobulin μbinding protein 2,IGHMBP2)基因,其发病主要与脊髓前角的α运动神经元受损有关,但至今详细的发病机制仍不明确[1-2]。大多数SMARD1患儿出生时四肢肌力、肌张力基本正常,常于出生后6周至6个月突然出现对称性四肢肌无力,远端重于近端、下肢重于上肢,膈肌麻痹合并呼吸窘迫,甚至呼吸衰竭需要呼吸机支持,也可出现外周神经感觉异常及自主神经功能紊乱。该疾病与其他婴幼儿期神经源性和肌源性疾病极相似,容易误诊或漏诊。而且该病预后差,大部分患儿死于1岁左右,仅极少部分患儿存活[3-5]。本文首次报道1例SMARD1患儿两个新的突变点,进行该疾病的家系分析,并结合相关文献,探讨诊断及治疗问题,以警示临床医生。
1临床资料
1.1发病情况 患儿 女,5个月,因“哭声弱5个月,双下肢乏力3个月余,呼吸困难15 d”于 2015年 4月9号入院。生后2 d,家长发现患儿哭声弱、吸吮无力,当时四肢可抬高,但生后6周出现四肢肌无力、足下垂(图1A),即到当地医院就诊,考虑脊髓性肌萎缩症 (spinal muscular atrophy,SMA),予激素、丙种球蛋白联合治疗,但效果欠佳,四个半月时开始出现呼吸窘迫,需要呼吸机辅助通气到广州市妇女儿童医疗中心住院。当时双上肢肌力近端3~4级,远端2级,双下肢肌力近端2级,远端 1级,均伴有肌张力低下。3个月时胸片提示右侧膈肌明显上抬(图2A)。4个月时头颅及脊柱MRI未见明显异常。染色体微阵列分析显示分别在染色体13q14.11和 14q32.33上出现400 Kb和462 Kb出现重排,但该重排未在数据库DGV,DECIPHER和 ISCA中找到,提示无明确的临床意义。肌肉活检提示肌纤维成束排列,部分肌纤维明显萎缩,考虑SMA (图3),但运动神经存活基因1 (survival of motor neuron 1,SMN1)缺失检测阴性。由于患儿原发病疑难复杂及反复撤机困难转入我院PICU。
患儿为G1P1,足月儿,顺产出生,过程顺利,出生体重3 kg,身长49 cm,父母体健,非近亲结婚。母孕期体健,家族史无特殊。
1.2体格检查 体温37.8℃,呼吸50次/min,脉搏128次/min,血压 98/69 mm Hg,身高 60 cm,体重 8 kg,头围 40 cm。带气管插管呼吸机转入,神志清,营养状态欠佳,精神运动发育均落后于同龄正常儿童。全身皮肤黏膜无皮疹、色素沉着,全身浅表淋巴结未扪及。全身汗多,额头、掌心为著。双侧眼睑无下垂,眼球运动无受限。双侧瞳孔等大等圆,对光反射灵敏。伸舌居中,舌体粗大,舌尖震颤。两侧鼻唇沟等深。颈软,无抵抗,颈肌无力,抬头困难。胸廓无畸形,双肺呼吸音粗,可闻及细湿性啰音。心律齐,各瓣膜听诊区未闻及杂音,无心包摩擦音。腹部平软,肝脾肋下未触及。双上肢肌力近端3级,远端2级;双下肢肌力近端2级,远端1级。四肢肌张力明显降低。双手指关节挛缩(图1B、C),余关节未见挛缩。双足下垂。双侧腹壁反射存在,膝反射弱,跟腱反射消失,踝阵挛阴性。双侧Oppenheim’s征 、Babinski’s 征 、Chaddock’s 征 和 Gordon’s 征 均 阴 性 ;Kenig’s征、Brudzinski’s征均阴性。
1.3辅助检查
1.3.1实验室检查 血常规、生化、肾功能、肝功能、体液免疫、凝血功能、乳酸、血尿氨基酸及有机酸、α-葡萄糖苷酶均正常,仅肌酸激酶(CK)及乳酸脱氢酶轻度升高。多次血气分析结果提示PO2<60 mm Hg。
1.3.2影像学检查 下肢平片提示小腿腓肠肌肌肉组织比正常明显减小(图2B)。
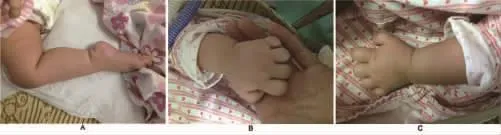
图1 患儿下肢和手。(A)下肢远端肌无力、足下垂;(B,C)手指挛缩
1.3.3心电图 窦性正常心率。
1.3.4肌肉活检病理 外院肌肉活检病理片送至我院病理科会诊,考虑SMA与先天性肌肉疾病相鉴别,建议基因检测(图 3)。
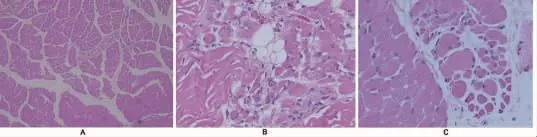
图3 腓肠肌的组织病理学图片。(A)HE染色 (100×);(B)HE染色 (200×);(C)HE染色 (400×),均可见肌肉纤维大小不一和萎缩,偶见玻璃变性,提示神经源性损害
1.4诊断与治疗 根据该患儿出生时四肢肌力、肌张力基本正常,于出生后6周突然出现对称性四肢肌无力,远端重于近端、下肢重于上肢,逐渐出现呼吸窘迫,最后呼吸衰竭需要呼吸机支持,胸片提示膈肌麻痹,腓肠肌病理提示神经源性损害,因此我们开始考虑SMARD1,患儿及其父母进行外显子基因测序,发现患儿IGHMBP2基因存在两个杂合突变点c.607G>C和 c.1418+5 G>A,患儿爸爸携带IGHMBP2基因的杂合突变点c.607G>C,患儿妈妈携带IGHMBP2基因的杂合突变点c.1418+5 G>A,但这两个突变点均未有报道,为了进一步证实,患儿父母的兄弟姐妹、患儿爷爷奶奶及外公外婆也进行外显子基因测序,发现患儿爷爷携带IGHMBP2基因的杂合突变点c.607G>C,患儿外公携带IGHMBP2基因的杂合突变点c.1418+5 G>A,结果可见,IGHMBP2基因的杂合突变点c.607G>C通过患儿爷爷遗传给患儿爸爸,IGHMBP2基因的杂合突变点c.1418+5 G>A通过患儿外公遗传给患儿妈妈,不幸的是,患儿遗传了其父母的这两个突变点而发病(图4),最后患儿11个半月大时因呼吸衰竭死亡。
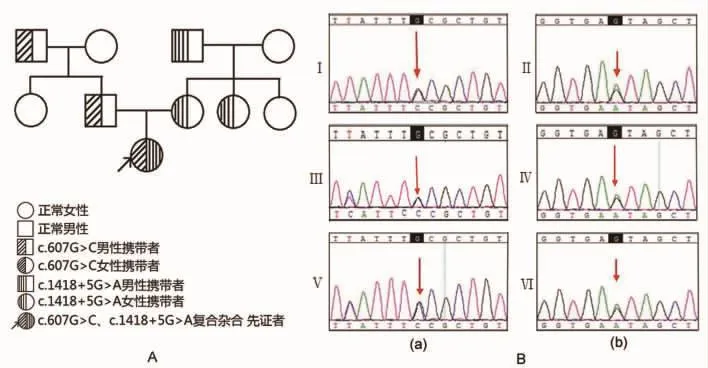
图4SMARD1患儿及其家庭成员的家系分析图和IGHMBP2基因测序结果。(A)SMARD1患儿及其家庭成员的家系分析图;(B)(a)错义突变c.607G>C;(b)剪接突变 c.1418+5 G>A。 I-爷爷,II-外公,Ⅲ-爸爸,Ⅳ-妈妈,Ⅴ和Ⅵ-患儿
2讨论
SMARD1是一种极其罕见的常染色体隐性遗传性疾病,典型的临床症状表现为出生体重低、哭声弱、吸奶无力、生长发育落后、对称性四肢肌无力(远端重于近端、下肢重于上肢)、腱反射消失、膈肌麻痹引起呼吸窘迫甚至呼吸衰竭等。包括典型的婴儿型和少年型SMARD1,大多数在婴儿时期发病,婴儿型SMARD1临床表现各异,从卧床不起到完全不需要轮椅活动自如的患儿[8]。目前仅有数例罕见的少年型SMARD1报道[9-10],相比婴儿型SMARD1,少年型SMARD1临床表现更轻些,迟发的呼吸衰竭和足下垂极少见,这可能与IGHMBP2蛋白酶的活性更高有关[8,11]。
本例患儿3个月时尚未需要呼吸机支持,当时胸部X线平片发现右侧膈肌比左侧抬高3个肋间隙,已提示膈肌麻痹征象,但容易被认为可能合并膈肌膨出,未引起临床医生的足够重视。虽未进行膈肌的肌电图检查证实膈肌麻痹,但回顾性分析本例,考虑患儿当时已可能发生膈肌麻痹。后转入我科,再结合患儿生后哭声弱、吸吮无力、远端重于近端的肢体肌无力、手指出现特征性的脂肪垫以及小腿X线平片提示小腿腓肠肌萎缩,考虑SMARD1,实施IGHMBP2基因检测,并进行家系分析,最终确诊为SMARD1。
SMARD1发病与IGHMBP2基因突变有关。IGHMBP2基因包含15个外显子、编码993个氨基酸和4个区域(腺苷三磷酸酶(ATP)区、单链核酸结合R3H域、DEXDc命名域和锌指区)[12-13]。研究表明IGHMBP2蛋白是个多功能蛋白,可影响多方面的细胞功能,包括转录、重组、复制、RNA核内编辑及细胞质内翻译[14-16]。虽该蛋白核心作用未完全明确,但研究提示大多数IGHMBP2致病突变基因削弱了其RNA依赖的ATP酶活性,除了c.1478 c>T(p.T493I)突变是降低IGHMBP2蛋白质稳态水平以外[11]。这说明了IGHMBP2致病突变基因通过降低酶活性或IGHMBP2稳态水平和减弱RNA解螺旋的能力而致病。寻找SMARD1基因突变点是为以后实施精准的基因治疗提供理论基础,因此,首先要完善SMARD1基因突变数据。SMARD1基因数据库(莱顿数据库)列出所有已经发表的IGHMBP2基因突变点,为遗传咨询提供有用信息(www.dmd.nl)。至今,全世界发现170多个IGHMBP2基因突变位点。IGHMBP2基因突变主要包括纯合子和杂合子突变,分布在IGHMBP2基因15个外显子,更常见位于外显子10和外显子12,而且在外显子12更容易出现c.1730 T>C(p.L577P)错义突变,在外显子10更容易出现c.1478C>T(p.Thr493Ile)错义突变和c.1488C>A(p.C496X)无义突变[17]。至今除IGHMBP2基因第四外显子,其余的外显子突变均有报道。本文患儿c.607G>C(p.A203P)错义突变位于IGHMBP2基因第五外显子,而c.1418+5 G>A剪切区突变点位于第九个内含子。至今这两个全新的突变点均未有报道,而我国仅报道过1例4岁10个月SMARD1女性患儿[18]。总结部分较有代表性文献(表1),发现大多数患儿出生后1个月至5个月突然出现对称性四肢肌无力,6个月内出现呼吸窘迫,绝大多数均出现膈肌上抬或麻痹,多数在1岁内死亡,有少部分存活,而存活患者肌无力及呼吸窘迫症状较轻。
那么,突变类型和表型之间有没有相关性?2001年ABDULAZI[19]首次在6个不同的SMARD1家庭中发现IGHMBP2基因突变与SMARD1的表型有关。有趣的是,SMARD1表型却存在很大的变异性。最近一项研究提示,在10例SMARD1患者中,存在纯合突变的患者比杂合突变表现出更严重的表型[20]。然而,关于突变类型和表型之间的关系至今尚未定论,而在本文表1中也没有发现它们的相关性。亦未在不同突变类型的患者中观察到临床的差异。
此外,值得注意的是,SMARD1患儿必须与婴幼儿期神经源性骨骼肌损伤引起的肌无力、呼吸窘迫、呼吸衰竭的其他疾病相鉴别。比如婴儿型脊髓性肌萎缩I型(SMAI型)[21],SMA伴脑桥小脑发育不全、SMA伴关节挛缩与骨折、SMA伴肌阵挛癫等。这些疾病的共同病理特点为脊髓的前角细胞受损,临床表现为四肢弛缓性瘫痪,肌电图提示骨骼肌的神经源性损害,但肌无力表现以肢体近端为著。此外,SMAI型患儿可检出SMN1第7、8外显子缺失;SMA伴脑桥小脑发育不全患儿头颅影像学检查可见特征性脑桥小脑发育不良;SMA伴关节挛缩与骨折患儿可见肢体长骨的多发性骨折;SMA伴肌阵挛癫患儿的脑电图显示特征性异常放大波。
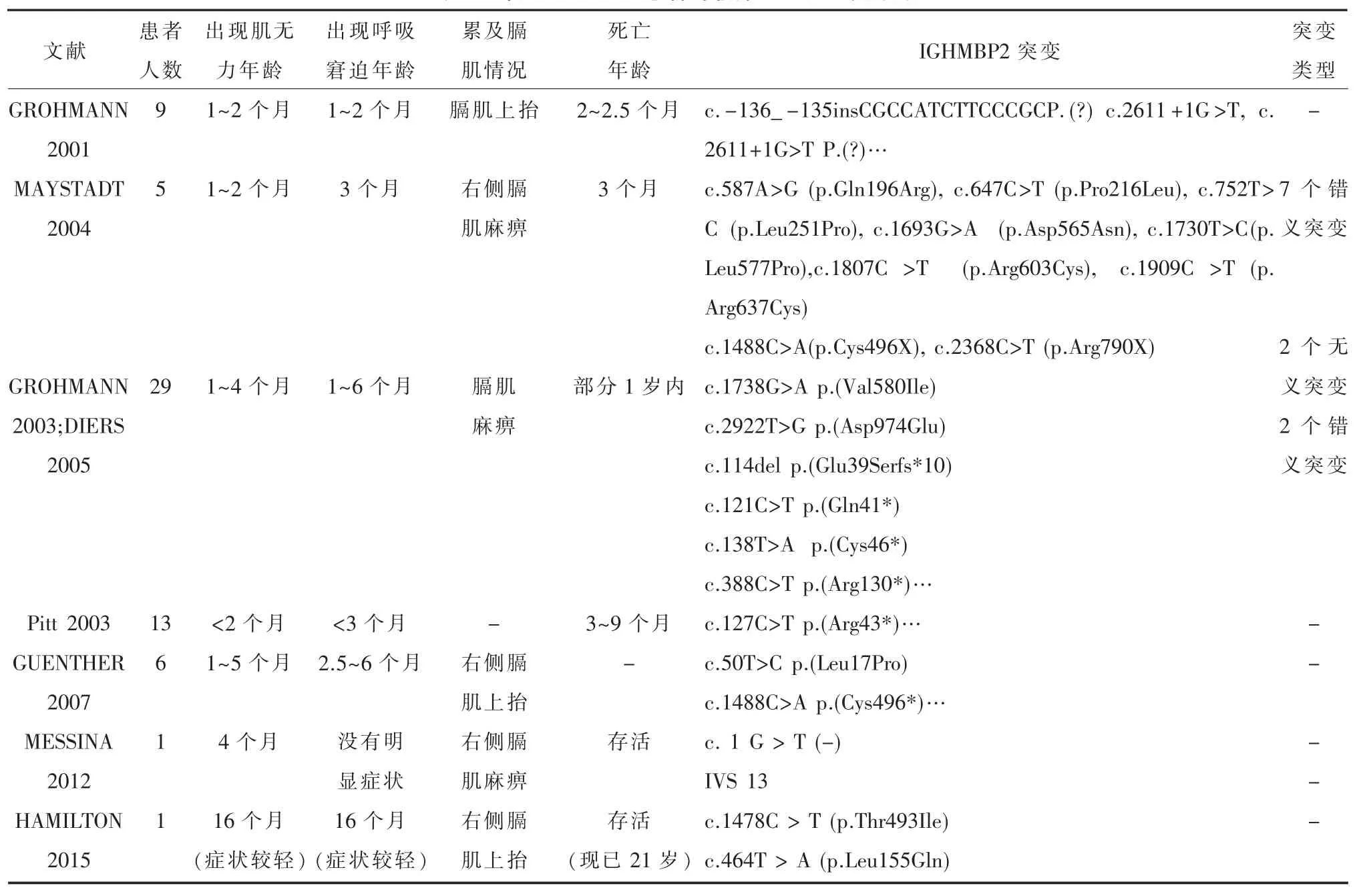
表1部分SMARD1患者的临床及基因突变数据
目前,SMARD1暂无有效的治疗策略,预后较差。IGHMBP2基因检测是早期诊断、治疗以及产前咨询、产前诊断的关键,及时处理呼吸窘迫、呼吸衰竭和预防呼吸道感染是延长SMARD1患儿生存期的重要办法。为更深入认识和治疗该罕见疾病,至今已投入很多临床前期研究。如基因靶向和干细胞治疗已取得较大的发展[22,23]。令人兴奋的是一些实验表明在症状出现前进行基因治疗更有效,而在症状出现后进行干细胞移植可发挥积极作用[17]。然而,这些临床前研究只是初步的,仍需更进一步地深入研究,尽早实现基因精准治疗。
[1]VANDERPOL WL,TALIM B,PITT M,et al.190 th ENMC international workshop:Spinal muscular atrophy with respiratory distress/distal spinal muscular atrophy type 1:11-13 May 2012,Naarden,The Netherlands [J].Neuromuscul Disord,2013,23(7):602-609.
[2]TACHI N,KIKUCHI S,KOZUKA N,et al.A new mutation of IGHMBP2 gene in spinal muscular atrophy with respiratory distress type 1[J].Pediatr Neurol,Apr;32(4):288-290.
[3]ECKART M,GUENTHER UP,IDKOWIAK J,et al.The natural course of infantile spinal muscular atrophy with respiratory distress type1(SMARD1)[J].Pediatrics,2012,129(1):148-156.
[4]APPLETON RE,HUBNER C,GROHMANN K,et al.Congenital peripheral neuropathy presenting as apnoea and respiratory insufficiency:spinal muscular atrophy with respiratory distress type 1(SMARD1)[J].Dev Med Child Neurol,2004,46(8):576.
[5]PIERSON TM,TART G,ADAMS D,et al.Infantile-onset spinal muscular atrophy with respiratory distress-1 diagnosed in a 20-year-old man[J].Neuromuscul Disord,2011,21(5):353-355.
[6]MELLINS RB,HAYS AP,GOLD AP,et al.Respiratory distress as the initial manifestation of Werdnig-Hoffmann disease[J].Pediatrics,1974,53(1):33-40.
[7]ZERRES K,RUDNIK SS.93rd ENMC international workshop:non-5q-spinal muscular atrophies(SMA)—clinical picture(6-8 April 2001,Naarden,The Netherlands)[J].Neuromuscul Disord,2003,13(2):179-183.
[8]MARK JH,CHERYL L,ANN OH.Growing up with spinal muscular atrophy with respiratory distress (SMARD1)[J].Neuromuscular Disorders,2015,25(2):169-171.
[9]GUENTER UP,HANDOKO L,VARON R,et al.Clinical variability in distal spinal muscular atrophy type 1(DSMA1):determination of steadystate IGHMBP2 protein levels infive patients with infantile and juvenile disease[J].J Mol Med,2009,87(2):31-41.
[10]KAINDL AM,Guenther UP,RUDNIK SS,et al.Spinal muscular atrophy with respiratory distress type 1(SMARD1)[J].J Child Neurol,2008,23(2):199-204.
[11]JEDRZEJOWSKA M,MADEJ P,FIDZIANSKA A,et al.Severe phenotypes of SMARD1 associated with novel mutations of the IGHMBP2geneandnucleardegenerationofmuscleand Schwann cells[J].Eur J Paediatr Neurol,2014,18(2):183-192.[12]MAJID A,TALAT K,COLIN L,et al.Heterogeneity in spinal muscular atrophy with respiratory distress type 1 [J].J Pediatr Neurosci,2012,7(3):197-199.
[13]ECKART M,GUENTHER UP,IDKOWIAK J,et al.The natural course of infantile spinal muscular atrophy with respiratory distress type 1(SMARD1)[J].Pediatrics,2012,129(1):148-156.
[14]LIEPINSH E,LEONCHIKS A,SHARIPO A,et al.Solution structure of the R3H domain from human Smubp-2 [J].J.Mol.Biol,2003,326(1):217-223.
[15]MIAO M,CHAN S,FLETCHER G,et al.The rat ortholog of the presumptive flounder antifreeze enhancer-binding protein is a helicase domain-containing protein [J].Eur.J.Biochem,2000,267:7237-7245.
[16]PLANELL S,SCHROEDER D,RODICIO M,et al.Biochemical and genetic evidence for a role of IGHMBP2 in the translational machinery[J].Hum.Mol.Genet,2009,18(12):2115-2126.
[17]FRANCESCA P,PAOLA R,FRANCESCA M,et al.The wide spectrum of clinical phenotypes of spinal muscular atrophy with respiratory distress type 1:A systematic review [J].Journal of the Neurological Sciences,2014,346(1-2):35-42.
[18]麦嘉卉,马伟科,韩春锡,等.脊髓性肌萎缩伴呼吸窘迫1型1例回顾性研究 [J].中国临床神经科学杂志,2015,23(3):280-286.
[19]ABDULAZIZ AL,SAMAN A,TOMOUM H.Infantile spinalmuscular atrophy with respiratory distress type 1:a case report[J].J Child Neurol, 2010,25(6):764-769.
[20]STALPERS XL,VERRIPS A,POLLTHE BT,et al.Clinical and mutational characteristics of spinal muscular atrophy with respiratory distress type 1 in The Netherlands[J].Neuromuscul Disord,2013,23(6):461-468.
[21]BAUGHN J,GERSHAN W,RAO A.Noisy breathing and hemidiaphragm paralysis progressing to respiratory failure in an infant[J].Pediatr Pulmonol,2011,46(8):817-819.
[22]CORTI S,NIZZARDO M,NARDINI M,et al.Motoneuron transplantation rescues the phenotype of SMARD1(spinal muscular atrophy with respiratory distress type 1)[J].J Neurosci,2009,29(38):11761-11771.
[23]CORTI S,LOCATELLI F,PAPADIMITRIOU D,et al.Transplanted ALDHhiSSClo neural stem cells generate motor neurons and delay disease progression of nmd mice,an animal model of SMARD1[J].Hum Mol Genet,2006,15(2):167-187.
描述统计检验结果需注意的问题
论文描述统计检验结果时,应注意以下问题:
1.P值指零假设成立的前提下,出现目前样本数据对应的统计量(如t值、F值、χ2值等)乃至更极端数值的概率。因此,描述统计检验结果时应给出统计量(如t值、F值、χ2值等)和P值。
2.目前统计分析软件已可以计算精确的P值,文中应报告精确的P值。当P值过小,统计软件输出结果P值为“0.000”,是因为目前小数位数不足以显示有效数字,在文中描述结果应写为“P<0.001”或“P<0.01”。
3.当P<0.05时,其统计学含义为可以拒绝零假设,因此可以得到“组间差异有统计学意义(significant difference)”的结论,而不译作“有显著差异”。更不能因为P值较小,如P<0.01,而称“差异非常显著”。
10.3969/j.issn.1002-0152.2017.09.013
☆ 2010年国家临床重点专科建设项目(编号:2011-872);广东省科技计划基金(编号:2013B021800276);广州市科技计划基金(编号:201510010148
* 中山大学附属第一医院PICU(广州,510080)
**中山大学附属第一医院神经内科(广州,510080)
○☆ 通信作者(E-mail:tangwen@mail.sysu.edu.cn)
2016-10-25)
李立)
猜你喜欢
保健与生活(2022年13期)2022-07-06
疯狂英语·新阅版(2021年8期)2021-09-10
种子(2021年3期)2021-04-12
中华养生保健(2021年18期)2021-02-13
世界科学技术-中医药现代化(2020年2期)2020-07-25
中国现代神经疾病杂志(2020年1期)2020-01-08
中国中医急症(2019年10期)2019-05-21
北京广播电视报(2019年8期)2019-03-27
武警医学(2019年2期)2019-03-05
校园英语·下旬(2017年7期)2017-07-14
