
分散固相萃取净化/高效液相色谱-串联质谱法测定水产品中的5种硝基咪唑类药物残留
2017-11-29 08:15:24王红青邱红钰
分析测试学报 2017年11期
励 炯,孙 岚,王红青,邱红钰,康 健
(1.杭州市食品药品检验研究院,浙江 杭州 310017;2.岛津技迩(上海)商贸有限公司,上海 200052)
分散固相萃取净化/高效液相色谱-串联质谱法测定水产品中的5种硝基咪唑类药物残留
励 炯1*,孙 岚1,王红青1,邱红钰1,康 健2
(1.杭州市食品药品检验研究院,浙江 杭州 310017;2.岛津技迩(上海)商贸有限公司,上海 200052)
建立了水产品中5种硝基咪唑类药物的高效液相色谱-串联质谱检测方法。样品经含0.1%氨水乙腈提取,加无水硫酸钠、C18-N以及NH2-PSA净化剂后涡旋振荡对样品进行净化,以Merck Chromolith Performance RP-18e(4.6 mm×100 mm)色谱柱分离,甲醇和0.1%甲酸水溶液为流动相梯度洗脱,正离子模式电喷雾电离,配合多反应离子扫描(MRM)定性定量分析目标化合物。考察了提取剂中氨水和净化剂的用量对加标回收率的影响,在优化实验条件下,5种硝基咪唑类药物在20~500 μg/L范围内线性关系良好,r2≥0.998 9;3个加标水平下的方法回收率为77.2%~94.8%;定量下限为0.7~2.0 μg/kg。该方法快速、简单、准确,适用于水产品中5种硝基咪唑类药物残留的检测。
高效液相色谱;串联质谱;分散固相萃取;硝基咪唑;水产品
硝基咪唑类药物(Nitromidazoles,NMZs)是一种具有5-硝基取代咪唑杂环结构的化合物(化学结构见图1),常见的主要有甲硝唑(Metronidazole,MNZ)、地美硝唑(Dimetridazole,DMZ)、洛美硝唑(Ronidazole,RNZ)等[1]。NMZs具有抗菌和抗原虫作用,在水产品养殖中广泛应用于寄生虫的防治与感染[2-3]。但由于NMZs含有的硝基杂环类化合物具有细胞诱变性,并且经生物体内代谢后形成的羟基甲硝唑(MNZOH,MNZ 的代谢物) 和羟基二甲硝咪唑(HMMNI,DMZ 和RNZ 的共同代谢物)同样具有原药类似毒性,其残留对动物源性食品安全构成了直接威胁,许多国家和地区已禁止该类药物用于食源性动物,我国也于2002 年禁用了该类药物[4-6]。
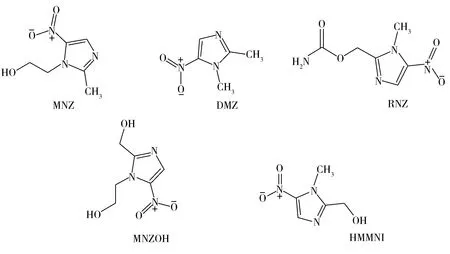
图1 MNZ、DMZ、RNZ、MNZOH和HMMNI的化学结构式Fig.1 Chemical structures of MNZ,DMZ,RNZ,MNZOH and HMMNI
目前国内外用于硝基咪唑类药物及其代谢物的检测方法主要有液相色谱法(HPLC)[7-9]、液相色谱-质谱联用法(LC-MS/MS)[10-12]、气相色谱-质谱法(GC-MS)[13-15]等。HPLC 法灵敏度低,易被杂质干扰,且无法准确进行定性;GC-MS方法繁琐,干扰因素多,适用范围有限;而LC-MS/MS 兼具灵敏度高及抗干扰性强的优势,广泛应用于硝基咪唑类药物多残留的检测。
前处理作为关键技术,直接影响方法灵敏度和精密度。液质联用前处理方法多以乙酸乙酯提取为主,用小柱净化,但其成本高、耗时长、步骤繁琐,操作不当易造成损失而影响定量准确性。本研究建立了分散固相萃取净化/高效液相色谱-串联质谱法检测水产品中硝基咪唑类药物及其代谢产物残留的方法,采用分散固相萃取技术进行前处理,省去了浓缩、固相小柱萃取等步骤,具有灵敏、简便、高效的优点,可满足水产品质量监测需求。
1 实验部分
1.1 仪器与试剂
美国Agilent公司高效液相色谱-串联四极杆质谱联用仪,配Agilent 1260高效液相色谱仪、Agilent 6410串联四极杆质谱仪,Milli-Q去离子水发生器(美国Millipore公司),MS3旋涡混合器。
甲硝唑(MNZ)对照品(批号:106700,Dr.Ehrenstorfer GmbH,含量99.8%);洛硝哒唑(RNZ)对照品(批号:101507,Dr.Ehrenstorfer GmbH,含量99.0%);地美硝唑(DMZ)对照品(批号:92632,Dr.Ehrenstorfer GmbH,含量99.0%);羟基甲硝唑(MNZOH)对照品(批号:281527,WITEGA公司,含量100%);羟甲基甲硝咪唑(HMMNI)对照品(批号:272032,WITEGA公司,含量99.5%);甲醇、乙腈(色谱纯,德国Merck公司);甲酸(95%,色谱级,美国Sigma公司);氯化钠、无水硫酸钠(分析纯);十八烷基键合硅胶吸附剂(C18-N)和NH2-丙基乙二胺吸附剂(NH2-PSA)(岛津技迩公司)。
1.2 色谱与质谱条件
色谱柱:Merck Chromolith Performance RP-18e(4.6 mm×100 mm);流动相:A为含0.1%甲酸水溶液,B为甲醇;梯度洗脱程序:0~4 min,5%~15%A;4~4.1 min,15%~5%A;4.1~7 min,5%A。柱温:30 ℃;流速:0.5 mL/min,进样量2 μL;运行时间:7 min。
离子源:电喷雾离子源(ESI),正离子检测方式(ESI+);多反应检测(MRM模式),MRM进行分段扫描:0~2.7 min,MNZOH;2.7~4.2 min,HMMNI、MNZ和RNZ;4.2~7 min,DMZ;干燥气温度:350 ℃;干燥气流速:10 L/min;喷雾器压力:200 kPa;毛细管电压:4 kV;校准方法:质量轴自动调谐校正;其他质谱分析参数详见表1。

表1 5种硝基咪唑类药物的质谱分析参数Table 1 MS parameters for 5 nitromidazoles
*quantitative ion pair
1.3 样品制备
提取:准确称取5.0 g鱼肉试样于50 mL聚丙烯离心管中,精密加入10 mL含0.1%(体积分数)氨水的乙腈溶液,充分均质提取5 min,加入2 g氯化钠和5 g无水硫酸钠,旋涡1 min,以5 000 r/min离心5 min,将有机相全部转入10 mL聚丙烯离心管中,待净化。
净化:在待净化样液中加入净化剂(内含1 000 mg无水硫酸钠,200 mg C18-N以及200 mg NH2-PSA),旋涡1 min混匀,以5 000 r/min离心5 min,精密取5 mL上清液转移至试管中,40 ℃水浴下氮吹至干,精密加入1 mL 0.1%甲酸水-乙腈(90∶10,体积比),旋涡混匀1 min溶解残渣,用0.22 μm微孔滤膜过滤。
1.4 标准溶液的制备
分别精密称取MNZ、DMZ、RNZ 以及两种代谢产物MNZOH及HMMNI标准品各约10 mg,分别用甲醇溶解转移至10 mL容量瓶中,用甲醇定容至10 mL,作为对照品储备溶液(质量浓度约为1 000 mg/L)。分别精密吸取上述5种标准储备溶液适当体积于10 mL容量瓶中,用甲醇稀释成质量浓度为1 000 μg/L的标准中间溶液。精密吸取适当体积的该中间溶液,配成20、50、80、100、200、500 μg/L的标准系列溶液。
2 结果与讨论
2.1 质谱条件优化
分别取MNZ、DMZ、RNZ、MNZOH及HMMNI质量浓度为1 mg/L的标准溶液用于质谱条件优化。在ESI离子源下,分别用正离子模式和负离子模式进行全扫描,结果发现5种化合物均在ESI+模式下响应最好,故本方法采用正离子模式进行检测。
在正离子模式下对5种硝基咪唑类化合物进行母离子扫描,扫描范围为m/z150~250,得到 [M+H]+峰。确定母离子之后,调节适当的裂解电压,使得各母离子一级扫描时响应最高。确定母离子及其裂解电压,继续进行子离子扫描,以得到最佳的二级质谱条件,经仪器自带的二级质谱参数自动优化程序,得到5种化合物的二级质谱优化参数(表1)。
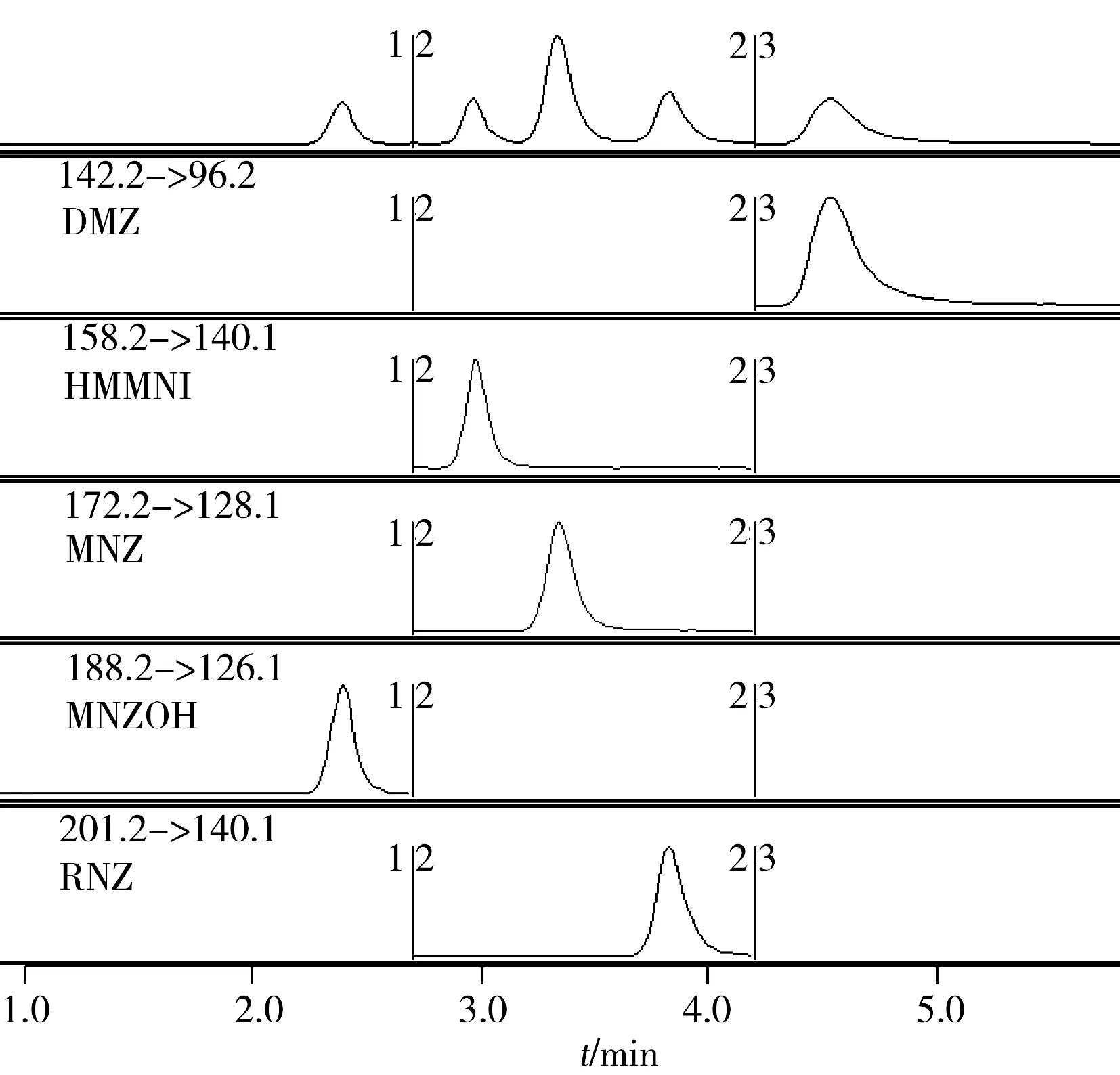
图2 5种硝基咪唑标准品的总离子图和MRM定量离子图Fig.2 TIC and MRM of five NMZs reference standards
2.2 色谱条件优化
本方法对Waters CORTECS-C18(2.7 μm,3.0 mm×50 mm)、Waters XBridge-C18(3.5 μm,3.0 mm×50 mm)、Merck Chromolith RP-18e(4.6 mm×100 mm)3种色谱柱的分离效果进行了比较。结果显示,普通的C18柱(CORTECS-C18和XBridge-C18)在分离MNZ和RNZ时比较困难,所以采用整体化色谱柱Merck Chromolith RP-18e进行分析。整体化色谱柱是利用极高纯度且不含金属的烷氧基硅胶为材料,采用新的液溶胶技术制备得到的整体化的多孔硅胶棒。这种硅胶棒具有典型的孔结构,即: 大孔结构和中孔结构,其中大孔的孔径为2 μm,相互交联成密集的大孔网络,保证流动相可以快速通过,却不会造成大的反压。填料骨架上13 nm的中孔保证了填料具有比颗粒型色谱柱更高的比表面积,从而可使该色谱柱在保证高柱效和低反压的条件下使用高流速,具有较好的色谱柱柱效。结果如图2所示,Merck Chromolith RP-18e对5种硝基咪唑类化合物的分离效果良好。
选择合适的流动相能直接影响目标化合物的分离、灵敏度等[16]。由于乙腈的极性高于甲醇,采用水相-乙腈体系作为流动相时,5种硝基咪唑类化合物的分离效果较差,分离度均未达到1.5,所以本研究采用极性相对较弱的甲醇作为有机相。在水相中加入一定比例的甲酸能促进离子化,因此本研究考察了不同含量的甲酸水溶液(0.1%、0.15%、0.2%)对5种目标化合物的分离效果,结果发现甲酸含量为0.1%时,5种目标化合物的分离效果最佳,且离子化程度较好,而当采用另外两个较高含量的甲酸水溶液时,MNZ和RNZ的分离度变差(<1.5)。因此本研究选择0.1%甲酸水溶液-甲醇为最佳的流动相。
2.3 提取及净化方式的优化
水产品中硝基咪唑类药物的提取溶剂一般采用乙酸乙酯、甲醇、乙腈以及一些混合溶剂,而分散固相萃取法提取兽药残留常采用的提取溶剂为含0.1%甲酸的乙腈溶液,所以本方法采用乙腈作为提取剂[17-18]。硝基咪唑类药物具有酸碱两性化学性质,在弱酸性条件下呈质子化状态,在弱碱性条件下呈游离分子状态,酸碱条件对MNZOH和HMMNI两种代谢产物的提取率影响较大,采用含0.1%甲酸的乙腈溶液作为硝基咪唑类药物的提取剂时,MNZOH和HMMNI的回收率均低于30%,其余3种硝基咪唑类药物的回收率为50%~60%,所以本文考虑在乙腈提取液中加入少量碱性物质来提高提取率。分别采用含0.05%、0.1%、0.15%氨水的乙腈溶液作为提取溶剂,结果显示,采用含0.1%氨水的乙腈时提取效率最高,5种硝基咪唑类药物的回收率均达到75%以上,并且氨水类挥发性碱性成分不会污染质谱离子源,符合实验要求。
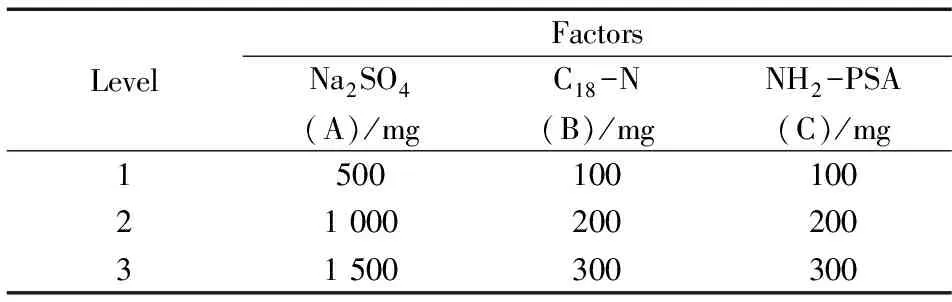
表2 无水硫酸钠、C18-N及NH2-PSA用量配比的正交试验因素水平Table 2 Orthogonal test factors and levels of the dosages of Na2SO4,C18-N and NH2-PSA
分散固相萃取常用的净化吸附剂为无水硫酸钠、C18-N、NH2-PSA,其中无水硫酸钠(一般用量为500~1 500 mg)主要用于除去样液中的水分,以保证其他吸附剂的吸附能力,C18-N(一般用量为100~300 mg)用于除去脂肪类杂质,NH2-PSA(一般用量为100~300 mg)主要吸附色素、金属离子、有机酸等。本研究通过对3种吸附剂的用量配比进行正交试验优化(无水硫酸钠、C18-N以及NH2-PSA用量分别作为因素A、B、C),每个因素取3个水平,按照L9(33)正交表试验,比较50 μg/L加标水平下各因素组合的回收率和净化效果,来选择最佳配比组合。正交试验因素见表2。正交试验结果表明,使用1 000 mg无水硫酸钠、200 mg C18-N以及200 mg NH2-PSA组成的净化剂可以获得良好的净化效果。
2.4 基质效应
基质效应会对方法灵敏度、精密度以及准确度造成影响[18]。MNZ、DMZ和RNZ在水产品中的基质效应不明显,但MNZOH和HMMNI存在一定的基质效应,本研究通过优化色谱分离条件较好地消除了基质效应,并采用相应基质配制标样进行校准,有效保证了定性、定量结果的准确、可靠。
2.5 线性关系与定量下限
取“1.4”配制的标准系列溶液,分别进样1 μL,以定量监测离子对离子流中谱峰峰面积(Y)为纵坐标,标准品的质量浓度(X,μg/L)为横坐标进行线性关系考察,结果见表3。5种硝基咪唑类药物线性关系较好,r2≥0.998 9。
取空白样品5 g,加入适当稀释的混合标准品溶液,按本方法制备供试品溶液分别进样测定,得5种硝基咪唑类药物的检出限(LOD,S/N=3)与定量下限(LOQ,S/N=10)分别为0.2~0.7 μg/kg和0.7~2.0 μg/kg,结果见表3。
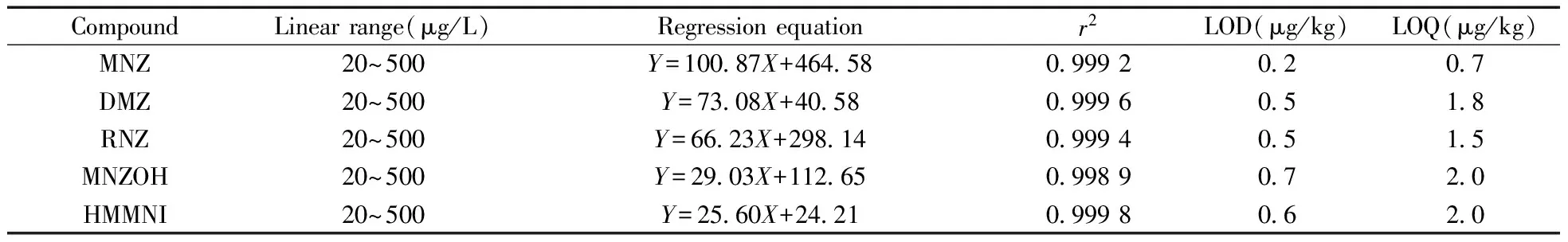
表3 5种硝基咪唑类药物的线性关系、检出限与定量下限Table 3 Linear regression results,LODs and LOQs of five NMZs
2.6 方法回收率与精密度
取空白样品,每份5 g,分别加入适量的混合对照品溶液,使供试品溶液最终加标水平分别为10、20、50 μg/kg,平行6份,按样品制备方法制备供试品溶液,检测含量并计算回收率,结果见表4。5种硝基咪唑类药物的平均回收率为77.2%~94.8%,相对标准偏差(RSD)为2.3%~7.8%。
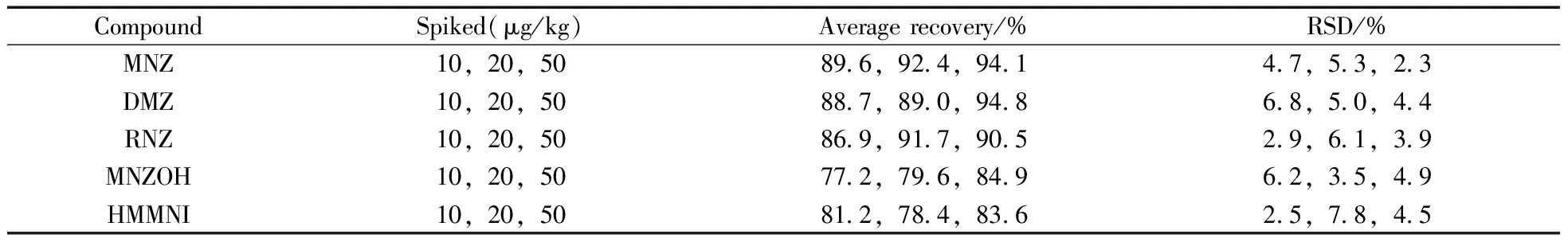
表4 平均回收率与精密度(n=6)Table 4 Average recoveries and precision(n=6)
2.7 实际样品检测
利用本研究建立的方法对杭州市市售的50批次水产品中5种硝基咪唑类药物进行检测,结果发现,50批次水产品中均未检出5种硝基咪唑类药物残留。
3 结 论
本研究建立了分散固相萃取净化/高效液相色谱-串联质谱测定水产品中硝基咪唑类药物及其代谢产物残留的分析方法,同时优化了色谱质谱条件以及提取和净化条件。该方法无需复杂的样品前处理,经含0.1%氨水的乙腈溶液提取,使用1 000 mg无水硫酸钠、200 mg C18-N以及200 mg NH2-PSA组成的净化剂进行净化吸附,即可有效降低基质效应,提高分析方法的选择性和灵敏度。
[1] Palermo A N,Reynoso A S,Nigro M L,Carballot M A,Mudry M D.BirthDefectsRes.A,2004,70(4): 157-162.
[2] Zhou J H,Shen J Z,Xue X F,Zhao J,Li Y,Zhang J Z,Zhang S X.J.AOACInt.,2007,90(3): 872-878.
[3] Mahugo-Santana C,Sosa-Ferrera Z,Torres-Padrón M E,Santana-Rodríguez J J.Anal.Chim.Acta,2010,665(2): 113-122.
[4] Lin Y Y,Su Y,Liao X L,Yang N,Yang X P,Choi M M F.Talanta,2012,88: 646-652.
[5] Ministry of Agriculture.No.235 Bulletin of the Ministry of Agriculture of the People’s Republic of China (农业部.中华人民共和国农业部公告第235号),2002.
[6] Ministry of Agriculture.No.193 Bulletin of the Ministry of Agriculture of the People’s Republic of China (农业部.中华人民共和国农业部公告第193号),2002.
[7] Gao X L,Wang D J,Wang J C,Zhang Z Y.J.HubeiUniv.:Nat.Sci. ( 高小龙,王大菊,汪纪仓,张泽英.湖北大学学报:自然科学版),2008,30(1): 71-75.
[8] Tan H Y,Yang G P,Yu P,Huang Y Y,Pei Q,Yuan H,Huang Z J,Xie F F,Mu L L.J.Pharmaceut.Biomed.,2013,71: 75-80.
[9] Wang Y,Zheng C Y,He F,Ye L H.FoodSci.(王杨,郑重莺,何丰,叶磊海.食品科学),2011,32(20): 197-199.
[10] Ding T,Xu J Z,Shen C Y,Jiang Y,Chen H L,Wu B,Zhao Z Y,Li G H,Zhang Q,Liu F.Chin.J.Chromatogr.(丁涛,徐锦忠,沈崇钰,蒋原,陈惠兰,吴斌,赵增运,李公海,张倩,刘飞.色谱),2006,24(4): 331-334.
[11] Liu Y M,Cao Y Z,Li J,Wu Y P,Zhang J J,Li X M,Ge N.Chin.J.Chromatogr.(刘永明,曹彦忠,李金,吴艳萍,张进杰,李学民,葛娜.色谱),2010,28(6): 596-600.
[12] Guo J,Ding L P,Wu W F,Zhao J H,Chen Z T.J.Instrum.Anal.(郭菁,丁立平,吴文凡,赵建晖,陈志涛.分析测试学报),2015,34(1): 28-34.
[13] Wang J L,Li Y K,Zhao J Y,Liu D C.Chin.J.Anal.Lab.(王金玲,李义坤,赵京杨,刘登才.分析试验室),2010,29(1): 107-110.
[14] Chen P R,Liu J N,Li X,He B.JiangsuAgric.Sci.(陈培荣,刘锦妮,李洵,何斌.江苏农业科学),2013,41(7): 292-293.
[15] He D,Li X Y,Xian Y P,Fang J,Guo X D.J.Instrum.Anal.( 何东,李秀英,冼艳萍,方军,郭新东.分析测试学报),2015,34(8): 911-916.
[16] Zhang L,Kong X H,Wang H,Li J H,He Q,Xu N S.Chin.J.Anal.Chem.(张璐,孔祥虹,王菡,李建华,何强,徐牛生.分析化学),2014,42(12): 1735-1742.
[17] Wei Y J,Zhu Z Y,Feng M,He J,Shen J R,He Z H,Qin X,Zhang L L,Qian Y P,Ding T.J.Instrum.Anal.(魏云计,朱臻怡,冯民,何健,沈金荣,何正和,秦娴,张伶俐,钱怡平,丁涛.分析测试学报),2017,36(3): 377-381.
[18] Zhang H W,Jian H M,Lin L M,Chen L Z,Liang C Z,Yuan T,Tang Z X,Cai X,Qin L Y.J.Instrum.Anal.(张鸿伟,简慧敏,林黎明,陈亮珍,梁成珠,袁涛,汤志旭,蔡雪,秦良勇.分析测试学报),2012,31(7): 763-770.
Determination of Five Nitromidazoles in Aquatic Products by High Performance Liquid Chromatography-Tandem Mass Spectrometry Combined with Dispersive Solid Phase Extraction
LI Jiong1*,SUN Lan1,WANG Hong-qing1,QIU Hong-yu1,KANG Jian2
(1.Hangzhou Institute for Food and Drug Control,Hangzhou 310017,China;2.Shimadzu-GL Sciences(Shanghai)Laboratory Supplies Co.,Ltd.,Shanghai 200052,China)
A high performance liquid chromatography-tandem mass spectrometric(HPLC-MS/MS) method was developed for the determination of five nitromidazoles in aquatic products.Samples were extracted with acetonitrile containing 0.1%(by volume) ammonium hydroxide,and then purified with a mixture of anhydrous sodium sulfate,C18-N and NH2-PSA.The analysis was performed by an HPLC-MS/MS system with Merck Chromolith Performance RP-18e(4.6 mm×100 mm) column.The mobile phase consisted of methanol and 0.1%(by volume) formic acid solution by gradient elution,and multiple reaction monitoring(MRM) mode with positive electrospray ionization was used.The effects of the dosages of ammonium hydroxide in extractant and anhydrous sodium sulfate,C18-N and NH2-PSA in cleaning-up agent were studied.Under the optimized analytical conditions,the calibration curves for five nitromidazoles were linear in the concentration range of 20-500 μg/L with their correlation coefficients(r2) not less than 0.998 9.The recoveries at three concentrations ranged from 77.2%to 94.8%with the LOQs of 0.7-2.0 μg/kg.The method is suitable for the determination of 5 nitromidazoles in aquatic products.
high performance liquid chromatography;tandem mass spectrometry;dispersive solid phase extraction;nitromidazole;aquatic product
2017-06-29;
2017-08-16
*
励 炯,硕士,副主任技师,研究方向:药品、食品、保健品检验,Tel:0571-85463893,E-mail:jokelee2@126.com
10.3969/j.issn.1004-4957.2017.11.011
O657.63;O657.7
A
1004-4957(2017)11-1357-06
猜你喜欢
中学生学习报(2021年20期)2021-11-24 15:30:52
鞍钢技术(2021年3期)2021-06-11 05:13:50
化工管理(2021年7期)2021-05-13 00:46:24
中国蜂业(2018年4期)2018-05-09 06:25:08
中国农资(2016年1期)2016-12-01 05:21:23
当代化工研究(2016年6期)2016-03-20 16:21:46
分析测试学报(2015年8期)2016-01-13 06:19:31
化工进展(2015年3期)2015-11-11 09:08:25
四川师范大学学报(自然科学版)(2015年3期)2015-02-28 14:08:00
无机化学学报(2014年8期)2014-02-28 17:32:46
