
超高效液相色谱-静电轨道阱质谱同时筛查定量饲料中的31种氨基甲酸酯类农药残留
2017-11-29 08:18:52吴剑平周悦榕李丹妮
分析测试学报 2017年11期
吴剑平,周悦榕,张 婧,李丹妮,严 凤,潘 娟
(上海市兽药饲料检测所,上海 201103)
超高效液相色谱-静电轨道阱质谱同时筛查定量饲料中的31种氨基甲酸酯类农药残留
吴剑平,周悦榕,张 婧,李丹妮*,严 凤,潘 娟
(上海市兽药饲料检测所,上海 201103)
建立了超高效液相色谱-静电轨道阱质谱法检测饲料中31种氨基甲酸酯类农药残留的检测方法和目标化合物的质谱数据库。饲料样品经100 mol/L HCl-甲醇(1∶3)提取,高速离心,经稀释后进样分析。采用Thermo Syncronis C18色谱柱(150 mm×2.1 mm,3 μm)作为固定相,0.1%甲酸和0.1%甲酸乙腈作为流动相进行梯度洗脱。质谱采用正离子扫描,以1对母离子和子离子的精确质量数进行定性,以母离子的峰面积进行定量分析。31种氨基甲酸酯类化合物在最优化条件下分离良好,在定量范围内线性良好,相关系数(r2)大于0.98,回收率达50.6%~109%,相对标准偏差(RSD)不大于8.9%。该方法具有简便、快速、灵敏、准确等特点,适用于饲料中氨基甲酸酯类化合物的同时定性筛查和定量分析。
氨基甲酸酯类农药;超高效液相色谱;静电轨道阱质谱;定性与定量;饲料
随着现代畜牧业养殖技术的不断进步发展,饲料的品质和安全越来越受广大养殖户的重视。针对饲料质量安全问题,为了杜绝饲料生产过程中的违禁物质添加现象,全国已开展了每年一度的跨省饲料抽样筛查工作,但对于饲料基质的谷物或油类作物中的农药残留尚未给予足够的重视。我国目前已发布的针对农药残留进行监管的主要法规公告包括:农业部公告194号(禁止11种高毒农药在果类作物上使用),农业部公告199号(禁止19种高毒农药用于蔬菜、果树、茶叶、中草药材),农农发〔2010〕2号文(禁止销售和使用23种高毒农药,限制19种农药在蔬菜、果树、茶叶、中草药材等作物上使用);农业部等5部门1586号公告(取消部分高毒农药的生产和使用许可);农业部193号公告和235公告(规定在动物可食组织中不得检出的农药种类),于2017年6 月正式实施的《中国食品安全国家标准:食品中农药最大残留限量》(GB 2763—2016),规定了433种农药在10大类食品中的4140项农药最大残留限量,成为我国农药残留食品安全市场监管的唯一强制性国家标准。综上所述,所有相关法规公告中对于谷物、油料作物、蔬菜、水果等食品中的农药残留监管十分有力,而对作为畜牧业重要投入品的饲料中的农药残留并没有明确规定,既未提出受监管的种类,也未提出相应的限量标准。国内对水果、蔬菜和谷物中多种农药残留检测方法的研究较多[1-3],而关于饲料中多种农药残留的检测方法却鲜见文献报道[4-5]。
目前,检测食品中多种农药残留的方法主要有高效液相色谱法(HPLC)、气相色谱法(GC)、色谱质谱联用法、生物传感器法和酶联免疫法等。气相色谱结合FPD或ECD检测器同时检测多种农药残留具有灵敏度高和特异性好的特点[6-7],但对于热不稳定和高沸点的化合物难以检测。液相色谱-串联质谱法可以较好地弥补这一缺点,同时也具有高灵敏度和高特异性的优点[8-9],有报道采用液相色谱-串联四极杆质谱法检测蔬菜、水果中65种农药残留[10],其检测灵敏度可达20 μg/kg。静电轨道阱质谱(Orbitrap mass)作为高分辨率质谱,相比串联四极杆质谱具有更高的质量精确度,同时也具有较高的灵敏度和较宽的线性范围,在多残留分析中可以兼顾较多质量数接近的化合物的定性准确性和浓度差别较大的化合物的定量精密性[11-13]。本研究利用静电轨道阱作为高分辨质谱的高灵敏度和高质量精度的特点,建立了31种常见氨基甲酸酯类化合物的色谱与质谱特征数据库和样品前处理方法,同时实现了对饲料样品中的氨基甲酸酯类农药残留的筛查、定性、确证和定量。该方法相比原有液质联用或气质联用方法大大提高了工作效率,方法的性能指标能达到国际标准,并为今后对饲料中农残化合物的预警监测和安全评估提供了技术保障。
1 实验部分
1.1 仪器与试剂
Ultimate 3000双三元超高效液相色谱、Excalibur控制软件、Trace Founder数据处理软件,Mass Frontier化合物结构推断软件,四极杆串联静电轨道阱质谱带HESI电离喷雾源(Q Exactive)、高速冷冻离心机(Allegra X-22R,Beckman Coulter);AL204电子天平(梅特勒-托利多仪器有限公司);水浴控温氮吹仪(N-EVAP,上海安谱公司)。C18分析柱(Syncronis C18,150 mm×2.1 mm× 3.0 μm,Thermo Fisher,美国);2,3,5-混杀威、3,4,5-混杀威、涕灭威、涕灭威亚砜、灭害威、恶虫威、丁酮威、丁酮威亚砜、丁酮砜威、甲萘威、克百威、3-羟基克百威、乙硫苯威、仲丁威、呋霜灵、异丙威、灭梭威、灭多威、速灭威、灭杀威、杀线威、抗蚜威、猛杀威、残杀威、特草灵、硫双威、久效威、灭除威、乙霉威、苯硫威、涕灭威砜(50 μg/mL,Bepure®,北京振翔科技有限公司);乙腈、甲酸(色谱纯,Merk公司,德国);去离子水(自制,电阻率≥18 MΩ·cm);高纯氮、高纯氩(纯度均为99.999%,上海佳杰特种气体公司)。
1.2 色谱条件
色谱柱:Syncronis C18(150 mm×2.1 mm×3 μm,美国Thermo公司)。流动相:A为0.1%甲酸,B为0.1%甲酸乙腈,采用梯度洗脱程序:0~1 min,5%B;1~8 min,5%~95%B;8~13 min,95%B;13.0~13.1 min,95%~5%B;13.1~17 min,5%B。柱温:30 ℃,流速:0.3 mL/min,进样量:10 μL。
1.3 质谱条件
使用Thermo Fisher公司的Q Exactive四极杆静电轨道阱质谱带HESI电离喷雾源质谱检测器对1 μg/mL的31种氨基甲酸酯类化合物标准溶液进行建库优化,兼顾各目标化合物优化后的质谱条件为:选择正离子模式,正电压3 200 V,鞘气压力344 kPa,辅助气流速5 L/min,吹扫气流速0.3 L/min,碰撞气压力0.2 Pa,毛细管温度 325 ℃,辅助气加热温度350 ℃,离子透镜射频电压50.0 V;扫描模式为全扫描加目标物二级离子碎片扫描模式,一级母离子扫描范围为m/z100~800,母离子扫描分辨率为70 000,静电轨道阱离子捕获器门限值(AGC Target)为3×106,最大注入捕获器时间为100 ms,二级碎片离子扫描分辨率为17 500,二级碎片离子捕获器门限值(dd-MS2 AGC Target)为1×105,最大注入捕获器时间(dd-MS2 Maximum inject time)为50 ms,同时最大响应离子(Top N)选取5个,动态排除时间5 s,数据处理软件提取离子质量精度为5 ppm,精确至小数点后5位。
1.4 定性与定量方法
按欧盟相关规定[14],以“1.2~1.3”的仪器方法,对目标农药进行标准品的进样并建库分析,获得最合适的目标物母离子精确质量数、保留时间以及其二级碎片离子的全扫描图谱,结合Mass Frontier所推断的子离子找出丰度最高的子离子和母离子一并作为确证所需的定性离子。以31种氨基甲酸酯类农药的保留时间、母离子精确质量数和子离子精确质量数相结合作为定性依据(表1)。定量时用一级母离子的精确质量数对总离子流图提取色谱,对目标物的色谱峰进行积分,通过曲线校准或单点校准进行定量分析。

表1 氨基甲酸酯类化合物的质谱分析条件Table 1 Mass spectrometry analysis conditions of carbarmates
1.5 样品前处理方法
准确称取饲料样品4.00 g,加入5.00 mL 0.1 mol/L盐酸溶液振荡湿润2 min,再加入15 mL甲醇,振荡2 min后室温下超声15 min,然后于8 000 r/min转速下离心5 min,取上层清液0.5 mL,加入0.1%甲酸0.5 mL混匀后14 000 r/min离心3 min,上清液经0.22 μm滤膜过滤后上机测定。
2 结果与讨论
2.1 定性定量方法依据
根据欧盟相关规定[14],以识别分(Identification points,IPs)为基础进行化合物的定性分析,对于不同法规要求的残留物,确证所需的IPs也不同,A类(违禁)需满足4 IPs,B类(限用)需满足3 IPs,农药残留适用违禁物质A类,因此需要4 IPs进行确证;不同的技术所获得的IPs也不同,静电轨道阱质谱作为高分辨质谱获得的1个母离子有2.0 IPs,相对应的1个子离子有2.5 IPs,因此建立目标化合物库时选出1对合适的母离子与子离子即可确证1个农残化合物。另外,化合物的元素组成决定其同位素峰的丰度比,因此一级和二级离子的同位素峰匹配分值也是重要的定性判断依据。由于静电轨道阱质谱的工作特性,其二级子离子精确质量数的分辨率(17 500)远低于一级母离子(70 000),特异性相对较差,且灵敏度远不如一级母离子的响应强度,因此本方法选择一级母离子的精确质量数为提取色谱质谱峰作为定量依据。
2.2 同分异构体的定性
本研究涉及的目标化合物包含多对同分异构体,比如丁酮威亚砜和涕灭威亚砜、丁酮威砜和涕灭威砜、丁酮威和涕灭威、灭除威和灭杀威等,它们具有相同的分子组成,因此即使静电轨道阱质谱拥有高达140 000分辨率的质量精度也无法区分这些化合物,但多残留筛查过程中不可排除这些化合物同时存在的可能,经实验验证,可采用3种不同方法解决这一问题∶①通过质谱前端的液相部分对元素组成相同但分子结构差异较大的化合物采用优化梯度洗脱条件、增加色谱分离度的方法,使同分异构体目标化合物的保留时间有较大差异,从而实现分离定性,例如本方法中丁酮威亚砜和涕灭威亚砜等;②通过质谱源区对同分异构体目标化合物母离子带电离行为的差异,分别检测其不同的加合物形式进行区分,例如将丁酮威和涕灭威,丁酮威砜和涕灭威砜这两对化合物经单标进样后发现,涕灭威和丁酮威砜在ESI+离子源作用下难以获得[M+H]+的母离子,而主要以[M+Na]+离子形式存在,而丁酮威和涕灭威砜母离子则主要以[M+H]+形式存在,因此在保留时间接近难以区分时,检测不同形式的母离子加合物也可以起到区分同分异构体的目的;③有些同分异构体化合物的保留时间非常接近,母离子加合物形式也相同,该情况下可通过单标进样后分析其所产生的特征碎片离子的差异进行区分。优化后所得典型特征离子的质量色谱(XIC)图见图1。
2.3 前处理方法的研究
考察了固相萃取法、分散固相萃取法和直接萃取后稀释进样法的前处理效果,结果发现固相萃取法和分散固相萃取法会对某些化合物产生歧视效应,因目标化合物的范围广泛,性质差异较大,因此选择直接萃取后稀释作为本方法的前处理方法。实验同时考察了0.1 mol/L盐酸溶液-甲醇(1∶3、1∶1、3∶1)以及0.1 mol/L盐酸溶液-乙腈(1∶3、1∶1、3∶1)对浓度为1倍定量下限(1LOQ)、2LOQ、5LOQ的31种氨基甲酸酯类化合物的提取效率,并将回收率结果在均值±40%以外的数据定义为乖离数据,统计其个数,实验结果见表2。结果发现使用0.1 mol/L盐酸溶液-甲醇(1∶3和1∶1)以及0.1 mol/L盐酸溶液-乙腈(1∶3)提取的回收率均值可达到70%以上,回收率较高;而使用0.1 mol/L盐酸溶液-甲醇(1∶3)提取时具有更小的歧视效应,因此选择其作为提取溶剂。

表2 饲料中31种氨基甲酸酯类化合物提取效率研究Table 2 Research on extraction efficiency of 31 carbarmates in feed
2.4 线性定量范围的确定
将31种氨基甲酸酯类化合物先用甲醇配制成50 mg/L的混合标准贮备液,再将空白饲料按“1.5”方法处理后获得空白样品基质溶液,用此溶液将标准贮备液分别稀释成1、2、5、10、20、50、100、500 μg/L标准曲线工作溶液,在优化条件下上机检测,获得各目标化合物的标准曲线,实验结果表明,在1~500 μg/L范围内31种化合物的线性良好,r2>0.98。
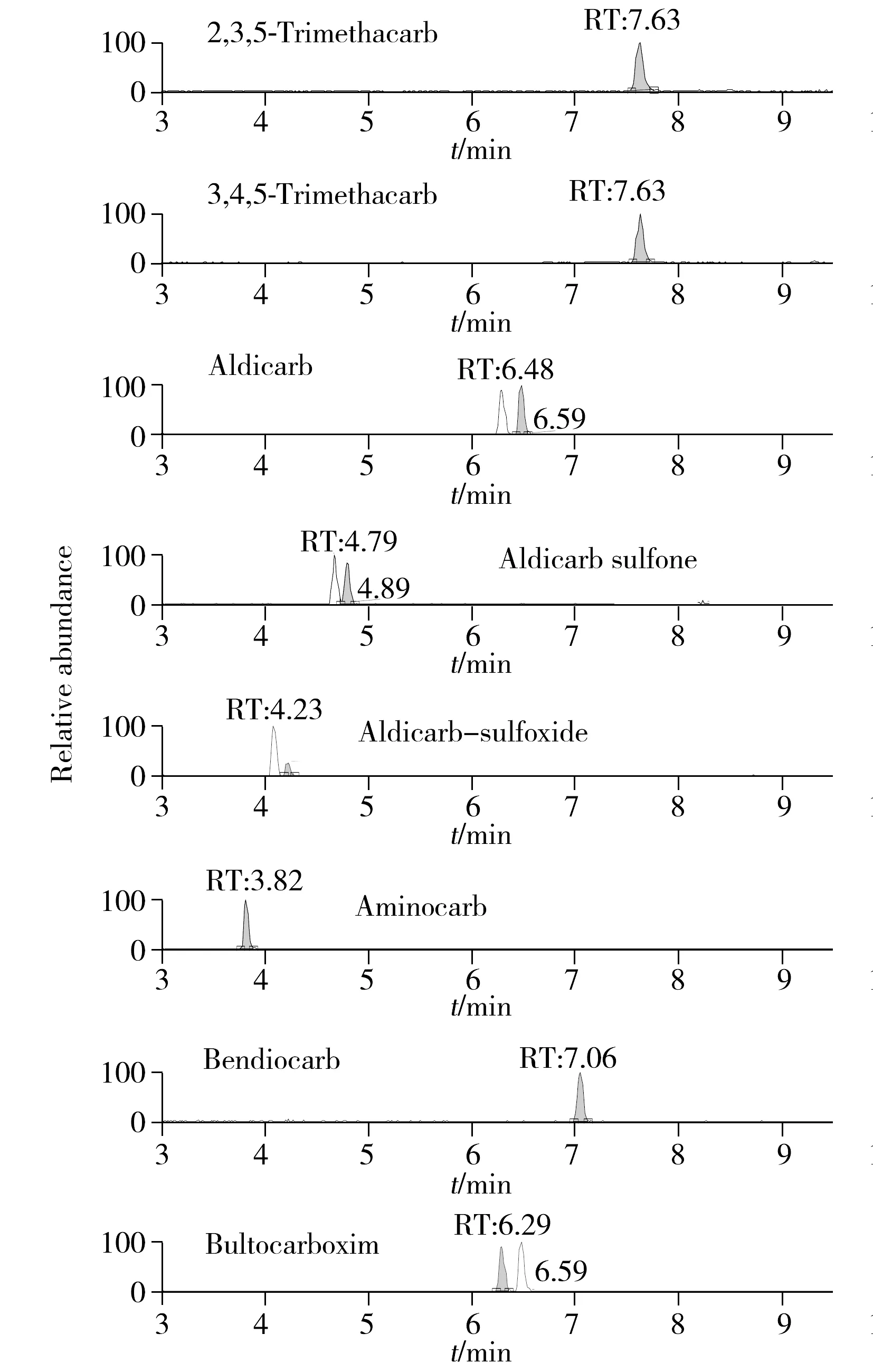
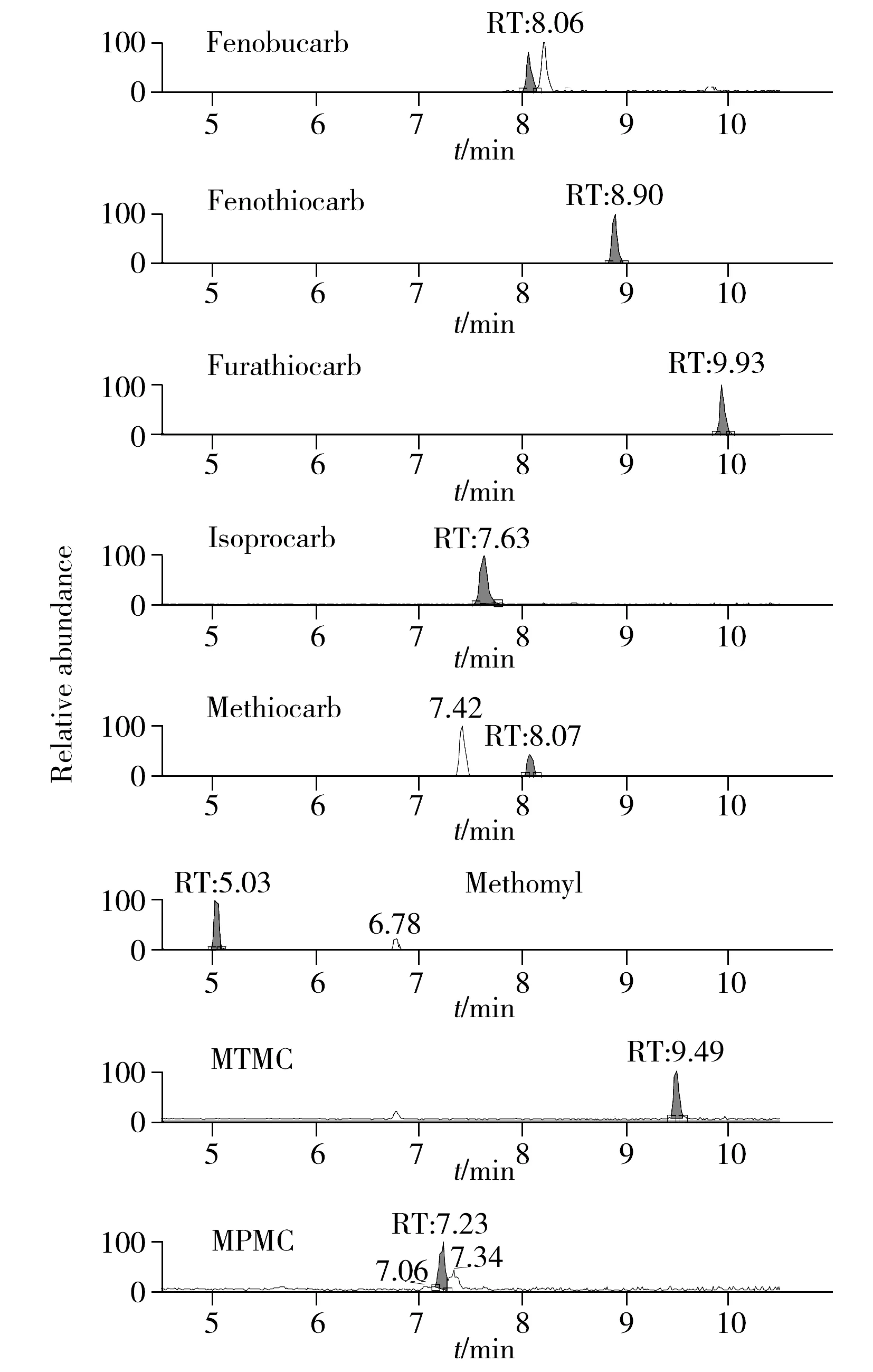

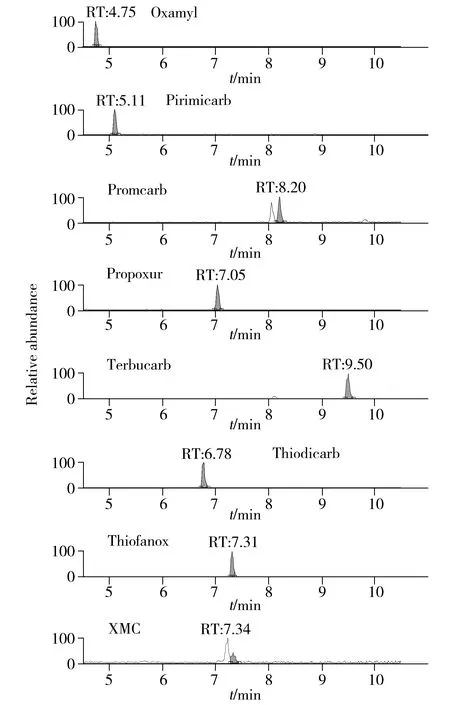
图1 定量下限浓度典型XIC图Fig.1 Typical XIC chromatograms of LOQ
2.5 检出限与定量下限
选择20个空白样品,按优化条件进行处理后上机测定,取与标准品图谱中相同保留时间的噪音信号平均值,以3倍信噪比(S/N≥3)为检出限(LOD),S/N≥10为定量下限(LOQ),确定该方法的LOD与LOQ,有关国家规定对不在其肯定列表目录中的农残化合物一律要求不得高于10 μg/kg的限量[15],因此本方法在结合信噪比和实际检测需要的情况下选择10 μg/kg作为方法检出限,20 μg/kg作为方法定量下限。实际样品检出限处的XIC图谱见图1。结果表明,检出限附近峰形良好,定性准确。
2.6 基质效应的考察
为验证方法基质效应,选取不同种类空白饲料6批,按“1.5”方法进行前处理,将其空白溶液中分别加入1LOQ的31种氨基甲酸酯类化合物标准溶液,测得其回收率为80%~120%,相对标准偏差(RSD)小于10%,证明该前处理方法的基质效应影响不大。
2.7 方法准确度与精密度
为验证方法精密度与准确度,采用标准添加法。使用6批不同饲料,每批称取等量6份,6份1组分3组,每组分别加入1LOQ、2LOQ、5LOQ的31种氨基甲酸酯类化合物标准溶液,结果见表3。实验结果表明,该方法检测饲料中31种氨基甲酸酯类化合物的回收率为50.6%~109%,RSD为3.9%~8.9%,证明该方法具有较高的准确度与精密度。
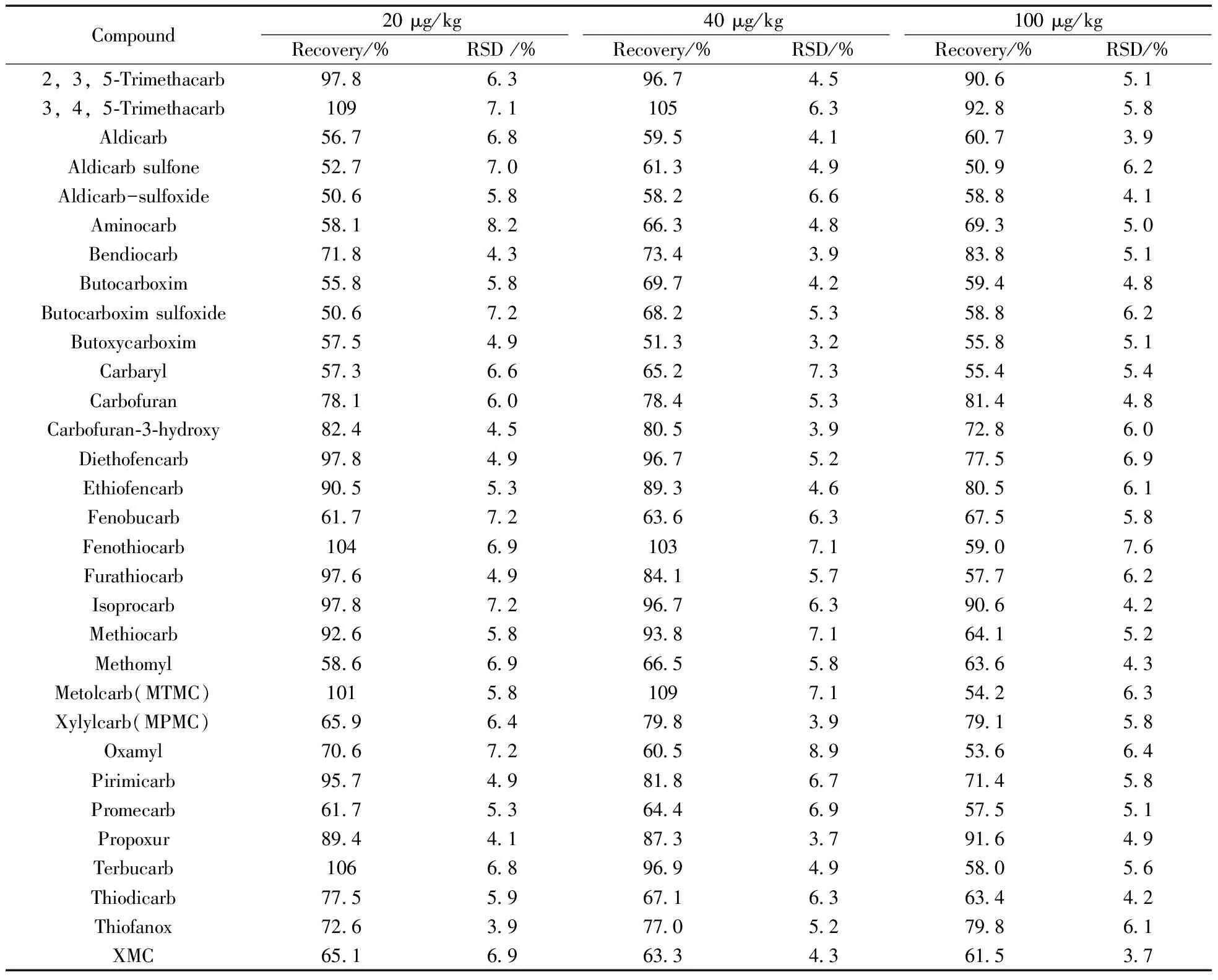
表3 31种氨基甲酸酯类化合物的加标回收率及相对标准偏差(n=6)Table 3 Recoveries and RSDs of 31 carbarmates in feed(n=6)
2.8 方法选择性的验证
选择20个空白样品,按优化条件处理后上机测定,未发现假阳性结果,表明该方法的选择性良好。
2.9 实际样品检测
取随机采集的某省份生产豆粕、玉米粉、玉米粒、谷糠、淀粉和玉米梗发酵物等饲料100批次,按本方法进行实际样品检测,将所得原始数据文件以Trace Founder数据处理软件进行筛查和定量处理,将筛查所得疑似阳性结果经保留时间,母离子精确质量数和同位素丰度比,以及子离子的精确质量数和丰度比进行确证。将确证后的阳性样品以外标标准曲线进行定量校准分析,结果如表4。据表4实验结果,该省份所抽取的100份样品中检出灭害威、苯硫威、乙霉威、3-羟基克百威和灭杀威,阳性率均未超过5%,其含量均远低于LD50(GB 2763-2016食品中农药最大残留限量),不会对生物造成毒害。但目前样本数量较少,所涵盖的农残化合物种类有限,在今后的工作中将进一步拓展采样数量和筛查范围。

表4 100批饲料的筛查和定量结果 Table 4 Screening and quantitation results for 100 feed
3 结 论
建立了超高效液相色谱-静电轨道阱质谱检测饲料中的31种氨基甲酸酯类农药的检测方法和目标化合物的质谱数据库。饲料样品经100 mol/L HCl-甲醇(1∶3)提取,高速离心,经稀释后进样分析。实验结果表明,31种氨基甲酸酯类化合物分离良好,在定量范围内均线性良好,相关系数(r2)均大于0.98,回收率为50.6%~109%,RSD不大于8.9%。本方法具有简便、快速、灵敏、准确等特点,适用于饲料中氨基甲酸酯类化合物的同时筛查和定量分析。
[1] Huang H Q,Rong D F,Cai C S,Xu Y,Zhu D Q.AcademicPeriodicalofFarmProductsProcessing(黄海泉,荣德福,蔡曹盛,徐颖,朱迪琦.农产品加工),2012,271(2):130-132,153.
[2] Liu B F,Liu G Y,Ma Y E,Yu C.BulletinofScineceandTechnology(刘宝峰,刘罡一,马又娥,余琛.科技通报),2010,26(1):93-99.
[3] Chen S B,Shan Z J,Hu Q H.FoodSci.(陈树兵,单正军,胡秋辉.食品科学),2004,12:152-155.
[4] Tekel J,Hatrik S.J.Chromatogr.A,1996,754:397-410.
[5] Gramt J,Rodgers C A,Chickering C D.J.AOACInt.,2010,93:1293-1301.
[6] Zhang W F,Hong Z T,Li J J.ChineseJournalofVeterinaryDrug(张卫锋,洪振涛,李嘉静.中国兽药杂志),2007,41(6):14-16.
[7] Zhang F,Zhang X Z,Luo F J,Chen Z M,Sun W J,Liu G M,Lou Z Y.J.Instrum.Anal.(张芬,张新忠,罗逢健,陈宗懋,孙威江,刘光明,楼正云.分析测试学报),2013,32(4):393-400.
[8] Wu J P,Zhang X,Gu X,Li D N,Yan F,Zhou Y R.J.Instrum.Anal.(吴剑平,张鑫,顾欣,李丹妮,严凤,周悦榕.分析测试学报),2013,32(7):834-839.
[9] Fan W J.DererminationofPesticideResiduesinFruitsandVegetablesbyDispersiveExtraction/HighPerformanceLiquidChromatography.Xinjiang:Shihezi University(樊雯娟.分散相萃取-高效液相色谱联用技术在果蔬农残中的研究与应用.新疆:石河子大学),2010.
[10] Wang X F.StudyonDeterminationof65PesticidesResiduesinSoil.Beijing:Chinese Academy of Agricultural Sciences(王小飞.土壤中65种农药残留检测技术的研究.北京:中国农业科学院),2013.
[11] Mi J B,Bi Y G,Zhao K X,Li S J,Xu H,He J,Zheng W J.J.Instrum.Anal.(宓捷波,毕玉国,赵孔祥,李淑静,许泓,何佳,郑文杰.分析测试学报),2015,34(11):1227-1232.
[12] Li M,Ma J C,Li H M,Xiong X C.J.Chin.MassSpectr.Society(李明,马家辰,李红梅,熊行创.质谱学报),2013,34(3):185-192.
[13] Liu H,Zhang Y L,Ding T,Wang S L,Xu N S,Gui Q W,Shen W J,Zhao Z Y,Wu B,Shen C J,Zhang R.J.Instrum.Anal.(柳菡,张亚莲,丁涛,王岁楼,徐牛生,桂茜雯,沈伟健,赵增运,吴斌,沈崇钰,张睿.分析测试学报),2014,33(5):489-498.
[14] 2002/657/EC.Commission Decision Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results,Text with EEA Relevance.Notified under Document Number C(2002)3044)(关于分析方法评价和结果解释执行委员会决议96/23/EC(于(2002)3044文件C中通报)欧盟委员会标准).
[15] G/SPS/N/JPN/145.The Final Draft of the Active List System of Agricultural Chemicals in Japan(日本农业化学品“主动列表”制度最终通报.日本厚生劳动省通报标准).
Simultaneous Screening and Quantitation of 31 Carbamate Pesticide Residues in Feed by Ultra Performance Liquid Chromatography-Orbitrap Mass Spectrometry
WU Jian-ping,ZHOU Yue-rong,ZHANG Jing,LI Dan-ni*,YAN Feng,PAN Juan
(Shanghai Municipal Supervisory Institute Veterinary Drugs and Feedstaff,Shanghai 201103,China)
To screen and quantify 31 carbamate pesticide residues in feed simultaneously,an ultra performance liquid chromatography-orbitrap mass spectrometric method and the target compounds data base were established.The feed sample was extracted with 100 mol/L HCl-methanol(1∶3).The extract was high-speedily centrifuged,purified with acid,then diluted and injected for analysis.Thermo Syncronis C18column(150 mm×2.1 mm,3 μm)was chosen as the solid phase with 0.1%formic acid and 0.1%formic acid acetonitrile as liquid phase by gradient elution.The mass spectrometer was operated in positive scan mode.One pair of parent ion and daughter ion were chosen for qualitation,and the response of the parent ion was used to achieve the quantitation.Under the optimal conditions,31 carbamates were separated well.Good linearities were obtained in their respective quantitative ranges with correlation coefficients(r2) more than 0.98.The average recoveries were in the range of 50.6%-109%and the relative standard deviations(RSDs) were not more than 8.9%.The method was convenient,economical,sensitive and accurate,and was applicable for the qualitation and quantitation of 31 carbamate pesticide residues in feed.
carbamate pesticides;ultra performance liquid chromatography;orbitrap mass spectrometry;qualitation and quantitation;feed
2017-06-29;
2017-08-04
上海市市级农口系统青年人才成长计划(沪农青字(2016)第2-5号)
*
李丹妮,硕士,高级畜牧师,研究方向:兽药安全评价及残留测定,Tel:021-62687816,E-mail:yure@sina.com
10.3969/j.issn.1004-4957.2017.11.012
O657.63;O657.7
A
1004-4957(2017)11-1363-07
猜你喜欢
毛纺科技(2021年8期)2021-10-14 06:51:16
食品安全导刊(2021年20期)2021-08-30 06:39:48
中成药(2018年12期)2018-12-29 12:26:08
中国有色金属学报(2018年2期)2018-03-26 07:58:48
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:36:55
当代化工研究(2016年5期)2016-03-20 16:21:35
合成化学(2015年1期)2016-01-17 08:55:47
中国塑料(2015年7期)2015-10-14 01:02:37
天然产物研究与开发(2014年7期)2014-04-27 14:16:08
特产研究(2014年4期)2014-04-10 12:54:22
