
X…Y(X=LiF,NH3,H2O; Y=HF,LiF)复合物中锂键、氢键的理论计算
2017-11-02 01:35刘雅萌高爱舫
华南师范大学学报(自然科学版) 2017年5期
刘雅萌, 李 俊, 高爱舫, 甄 岩
(1. 北京工业大学生命科学与生物工程学院, 环境与病毒肿瘤学北京市重点实验室, 北京 100124;2. 广东第二师范学院化学系,广州 510303; 3. 河北地质大学水资源与环境学院, 石家庄 050031;4. 北京工业大学环境与能源工程学院, 北京 100124)
X…Y(X=LiF,NH3,H2O; Y=HF,LiF)复合物中锂键、氢键的理论计算
刘雅萌1, 李 俊2, 高爱舫3, 甄 岩4*
(1. 北京工业大学生命科学与生物工程学院, 环境与病毒肿瘤学北京市重点实验室, 北京 100124;2. 广东第二师范学院化学系,广州 510303; 3. 河北地质大学水资源与环境学院, 石家庄 050031;4. 北京工业大学环境与能源工程学院, 北京 100124)
在CCSD(T)/cc-pVTZ水平下,对X…Y(X=LiF、NH3、H2O; Y=HF、LiF)复合物的9个结构进行几何构型优化和红外振动频率计算. 根据定域化分子轨道、原子自然电荷、Wiberg键级的分析表明HFLiF分子中的H—F键是共价键,而Li—F键则为离子键而非共价键. H3N…Y (Y=HF、LiF)、H2O…Y (Y=HF、LiF)中的氢键或锂键源于静电相互作用,并非共用电子的共价键. 结合能的计算表明: 与HF相比,LiF与X (X=LiF、H2O、NH3)的结合能更高;结合能从高到低依次为 LiF > NH3> H2O. 红外振动频率分析表明HF与NH3、H2O形成红移氢键,即 H—F键长增加,相应的H—F伸缩振动频率降低. H3N…LiF的Li—F键键长增加同时伸缩振动频率减少. 而LiF与H2O形成锂键后,键长增加0.001 6 nm,而Li—F的伸缩振动频率反而增加了2 cm-1,即蓝移锂键.
X…Y(X=LiF,NH3,H2O; Y=HF,LiF)复合物; 蓝移锂键; 氢键; 定域化分子轨道
Keywords: X…Y(X=LiF,NH3,H2O; Y=HF,LiF) complexes; blue-shifting lithium bond; hydrogen bond; localized molecular orbital
氢键在化学、物理和生物等领域中发挥着关键的作用,其成键规律及性质一直是研究热点[1-10]. 研究发现,在X—H…Y的氢键中,由于Y的孤对电子可以填入X—H的反键轨道形成电荷转移导致X—H键被削弱,体现为X—H的键长增加,且其伸缩振动频率降低,即红移现象[2-3]. 1998年AND等[4]在研究苯二聚体中的氢键时发现了与上述情况相反的蓝移现象,即X—H键长缩短且其振动频率升高而非降低. 蓝移氢键的发现引起了科学家的广泛兴趣[4-7]. 对于氢键或者锂键产生蓝移的原因,近年来人们试图从静电吸引作用和排斥作用[6]、相互竞争的吸引作用[7]、价键理论[3]进行解释,但尚未得到一致的结论. 锂及其化合物是重要的电池电极材料[11-12]. 锂与氢类似,只有1个价电子,这使得它可以与其它原子形成单键,如HF或LiF. 1970年,KOLLMAN,LIEBMAN和ALLEN等提出锂键后,锂键也成为化学领域的研究热点之一[13-14]. 与氢键相似,在锂键连接的X—Li…Y中,由于Y的作用导致X—Li的振动频率降低,表现为红移[14]. FENG等[15]在MP2/6-311++G(d,p)水平下对一系列含锂键复合物进行理论研究,发现F3C—Li…NH3中C—Li键长虽然增加了0.002 nm,但其振动频率增加了约100 cm-1,即蓝移锂键.
锂键、氢键都是分子间的弱相互作用. 在讨论化学键及其成键方式时,常用X…Y 复合物(X=N2、NH3、H2O等孤对电子供体; Y=LiF、HF、HLi等)作为研究对象[13].最近SHAHI和ARUNAN[16]在MP2/aug-ccpVTZ的水平下对上述复合物进行了理论研究,以讨论不同X、Y间形成的锂键或氢键,并预测了复合物的结合能、红外光谱等性质. 本文选择LiF、NH3、H2O分别与LiF、HF形成的复合物为研究对象,采用更为精确的研究方法进行理论计算,并结合定域分子轨道方法讨论其中氢键、锂键的成键方式和特点. 研究提供了更准确的结合能以及红外光谱结果,为研究含氢键、锂键化合物的实验研究提供一些理论线索.
1 计算方法
在CCSD(T)/cc-pVTZ水平下[17-18],对本文讨论的所有复合物进行优化得到其平衡几何构型. 在相同水平下,对所有构型进行谐振频率计算以获得振动频率和零点校正能(ZPVE). 本文采用wB97X-D 方法[19]结合cc-pVTZ基组计算所有构型的自然原子电荷(Natural atomic charge)、Wiberg键级[20],并用定域化分子轨道讨论了其中的化学键的性质. 所有的密度泛函理论计算均用Gaussian 09程序[21]完成;CCSD(T)计算由CFOUR程序[22]完成;自然原子电荷以及Wiberg键级由Gaussian09软件包中的NBO 3.1程序计算获得;定域化的分子轨道(LMOs)采用Pipek-Mezey[23]的方法,通过对正则分子轨道(CMOs)进行酉变换得到.
2 结果与讨论
2.1 HF、LiF、NH3和 H2O单体的计算
首先对HF、LiF、NH3、H2O分子进行计算研究. 优化得到的H—F键长为0.091 6 nm,其伸缩振动频率ν(HF) 为4 162 cm-1,强度为104 km/mol. NBO计算得到H—F Wiberg键级为0.71,原子自然电荷为0.54(H),-0.54(F). Li—F键长为0.157 7 nm,其伸缩振动频率ν(LiF) 为 907 cm-1,强度为142 km/mol. Li—F键的Wiberg键级为0.18,Li和F原子的原子自然电荷分别为0.91、-0.91. 计算得到的H2O分子中O—H键长为0.095 8 nm,H—O—H键角104°. NH3中N—H键长0.101 4 nm,键角113°. 上述构型的几何参数与实验数据基本一致[24-26],表明计算方法合理可靠.
2.2 X…Y复合物
优化得到的9个X…Y (X=LiF,NH3,H2O;Y= HF,LiF)复合物的几何构型(图1)及能量最低异构体的相关参数(表1).
HFLiF分子得到环型结构1和直线型结构2. 结构1的能量比结构2的低63 kJ/mol. 直线型结构2含有2个虚频,其数值均为218i cm-1. 沿着这2个虚频的振动方向进一步优化发现结构2变为环型结构1. 这表明结构2不是一个稳定的异构体.
与HFLiF相似, LiFLiF也得到环型结构3和直线型结构4. 环型结构3的能量比直线型结构4低117.7 kJ/mol. 直线型结构4的振动频率计算结果没有发现虚频,说明LiFLiF的直线型结构是稳定的异构体.
计算发现了2个能量接近的H3N…LiF结构, 其中对称性为C3v的结构5比结构6能量低约0.6 kJ/mol. 2个结构的频率计算表明其均为稳定的异构体. 具有2个能量相近的异构体表明H3N…LiF沿F(6)—Li(5)…N(1)键角折叠时所需能量很小,即H3N…LiF的锂键不具有固定的方向性. 而H3N…HF的优化计算只得到C3v的结构7,与结构6相似的H3N…HF结构在优化过程中转变为C3v的结构7. 表明氢键结合的H3N…HF具有一定的方向性. H2O…LiF的结构8为C2v对称性,频率计算证明其无虚频是稳定的异构体. 而C2v对称性的H2O…HF却存在1个约220i cm-1的虚频,沿着虚频的振动模式继续优化得到Cs对称的结构9.

图1 在CCSD(T)/cc-pVTZ水平下, X…Y (X=LiF,H3N,H2O; Y= HF,LiF)复合物的几何结构
Figure 1 Optimized geometry (bond length in nm, angle in degree) for X…Y (X=LiF,H3N,H2O; Y= HF,LiF) complexes obtained at CCSD(T)/cc-pVTZ level
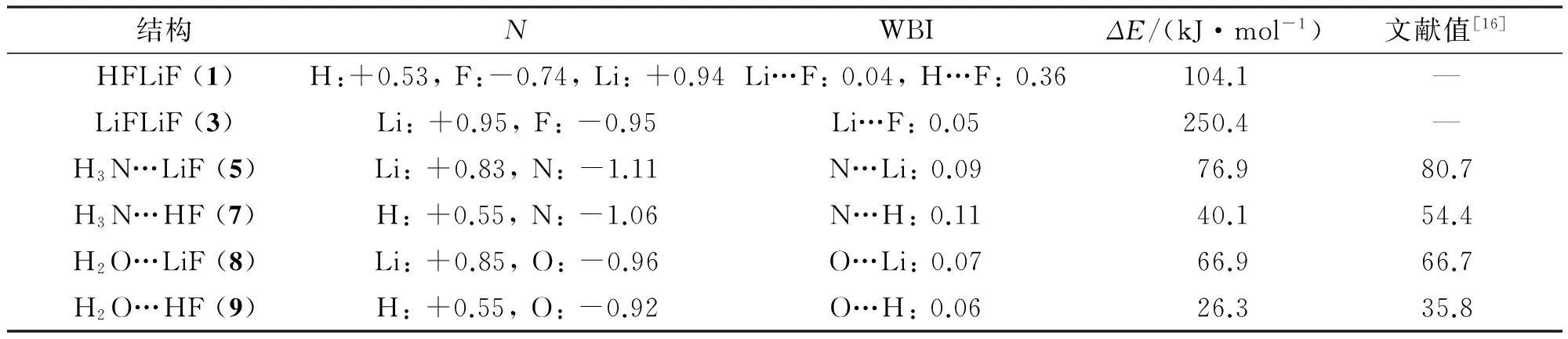
表1 不同结构零点能校正的结合能、原子自然电荷N以及Wiberg键级WBITable 1 Binding energies with ZPVE (ΔE), natural atomic charge (N), and Wiberg bond index (WBI) for different structures
2.3 原子自然电荷、Wiberg键级和定域化轨道
在X…HF(X=LiF,NH3,H2O)的3个能量最低异构体(表1)中,氢键的键级依次为0.06(9,O…H)、0.11(7,N…H)、0.36(1,F…H). 原子自然电荷的数据表明,H原子的电荷值范围0.53~0.55,与其形成氢键的N、O、F原子的自然电荷分别为-1.06、-0.92、-0.74. 可见,除HFLiF中的F…H键外,H3N…HF、H2O…HF中的氢键都以静电相互作用为主,共价键成分较少. 由图2可知,HFLiF中存在共享电子的共价键H—F,而H3N…HF、H2O…HF中仅能找到N、O原子的孤对电子轨道,没有形成共享电子的共价键.
采用与氢键相同的分析方法,X…LiF(X=LiF,NH3,H2O)中锂键的Wiberg键级均≤0.09. 原子自然电荷的数据,Li原子的电荷值范围0.85~0.95,与之形成锂键的N、O、F原子的自然电荷分别为-1.11、-0.96、-0.74. 可见,上述锂键都以静电相互作用为主,不存在共价键成分. Li原子与F、O、N原子间没有形成共享电子的共价键(图2).
2.4 结合能
复合物的结合能不仅可以反映连接复合物的相互作用的强度, 而且体现了复合物的稳定程度. 从表1可见,LiFLiF的结合能最高(250.4 kJ/mol), 而H2O…HF的结合能最低(26.3 kJ/mol). 其中含锂键的复合物(1,5,8)的结合能范围66~250 kJ/mol,结合能LiFLiF> LiFNH3> LiFH2O. 含氢键的复合物结合能分别 26.3、40.1 kJ/mol. 因此,锂键复合物的结合能高于氢键. 与H2O相比,与NH3形成的锂键或氢键更强. 文献[17]在MP2/aug-cc-pVTZ水平下获得的结合能与本文的数据相比,除H2O…LiF (8)基本一致外, MP2方法获得的其他复合物的结合能偏高3~14 kJ/mol.

图2 X…Y复合物的能量最低异构体的定域分子轨道
2.5 红外振动光谱
在CCSD(T)/cc-pVTZ水平下得到的结构5、6、7、8、9的Y—F (Y=Li,H)键长及其变化量、Y—F (Y=Li,H)伸缩振动频率及其变化量列于表2. HF与NH3、H2O形成氢键后,H—F键长分别增加了0.002 9、0.001 6 nm,相应的H—F伸缩振动频率降低了359、644 cm-1,发生红移. LiF与NH3形成锂键后,Li—F键长增加了0.002 6 nm(结构5)、0.003 9 nm(结构6),频率则分别减少了4、78 cm-1,属于红移锂键. LiF与H2O形成锂键后(结构9),键长增加了0.001 6 nm,而Li—F的伸缩振动频率增加了2 cm-1出现蓝移的现象. 此结论纠正了前人在MP2/aug-cc-pVTZ水平下得到的H2O…LiF复合物LiF键伸缩振动频率红移19 cm-1[16]的结论.
表2在CCSD(T)/cc-pVTZ水平下得到的不同结构的X—F (X=Li,H)键长r及其Δr、Y—F (Y=Li,H)伸缩振动频率ν及其Δν
Table 2 Bond lengthsrof X—F (X=Li,H), theΔr, Y—F(Y=Li,H) stretching frequencies ν and theΔνfor different structures determined at CCSD(T)/ cc-pVTZ
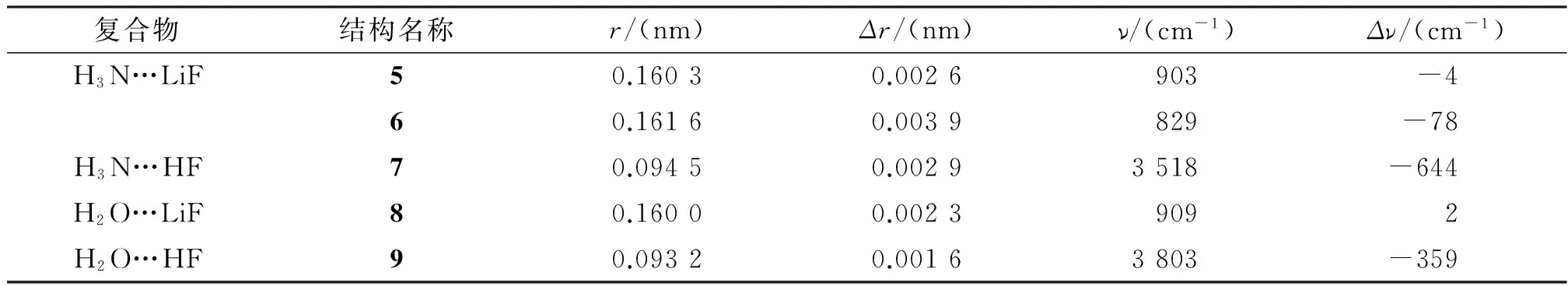
复合物结构名称r/(nm)Δr/(nm)ν/(cm-1)Δν/(cm-1)H3N…LiF50.16030.0026903-460.16160.0039829-78H3N…HF70.09450.00293518-644H2O…LiF80.16000.00239092H2O…HF90.09320.00163803-359
3 结论
自然原子电荷、Wiberg键级以及定域轨道分析表明, 对X…Y (X=LiF,NH3,H2O;Y=HF,LiF)复合物中的锂键来自于静电相互作用,可视为离子键而非共价键. 除HFLiF存在共价键成分的氢键外,其余复合物中的氢键也以静电相互作用为主.
获得了较精确的结合能, 表明前人采用MP2方法计算的结果除H2O…LiF外偏高3~14 kJ/mol. 含锂键的复合物结合能大小依次为LiFLiF>LiFNH3>LiFH2O. 含氢键的复合物的结合能HFNH3>HFH2O. 此外,LiFNH3、LiFH2O的结合能比HFNH3、HFH2O的高,表明锂键的强度比氢键更高.
HF与NH3、H2O形成氢键后,H—F键长增加,相应H—F伸缩振动频率降低(红移). LiF与NH3形成锂键后,Li—F键长增加、频率降低,产生红移. 而LiF与H2O形成锂键后,键长增加0.001 6 nm,而Li—F的伸缩振动频率增加2 cm-1,表现为蓝移. 这纠正了前人关于复合物H2O…LiF中Li—F键伸缩振动频率红移的结论.
[1] DESIRAJU G,THOMAS S. The weak hydrogen bond[M]. Oxford:Oxford University Press,2001.
[2] RATAJCZAK H. Charge-transfer properties of the hydrogen bond. I. Theory of the enhancement of dipole moment of hydrogen-bonded systems[J]. Journal of Physical Chemistry 1972,76:3000-2004.
[3] CHANG X,ZHANG Y,WENG X,et al. Red-shifting versus blue-shifting hydrogen bonds:a perspective from ab Initio valence bond theory[J]. Journal of Physical Chemi-stry A,2016,120(17):2749-2756.
[5] 杨颙,张为俊,裴世鑫,等. N-H…O红移氢键和蓝移氢键的理论研究[J]. 中国科学B,2006,36(3):218-226.
[6] LI X S,LIU L,SCHLEGEL H B. On the physical origin of blue-shifted hydrogen bonds[J]. Journal of the American Chemical Society,2002,124(32):9639-9647.
[7] SCHUSTER P,ZUNDEL G,SANDORFY C. Thehydrogen bond:recent developments in theory and experiments:structure and spectroscopy[M]. Amsterdam:North-Holland,1976.
[8] JEFFREY G A,SAENGER W. Hydrogenbonding in biological structures[M]. Berlin:Springer Science & Business Media,2012.
[9] SCHEINER S. In Theoreticalmodels of chemical bonding[M]. Berlin:Springer-Verlag,1991.
[10] AND J J,JEMMIS E D. Red-,blue-,or no-shift in hydrogen bonds: a unified explanation[J]. Journal of the American Chemical Society,2007,129(15):4620-4632.
[11] 赵瑞瑞,杨子莲,杜鹏,等. 锂源对固相合成LiNi0.5Co0.2Mn0.3O2正极材料性能的影响[J]. 华南师范大学学报(自然科学版),2015,47(2):48-52.
ZHAO R R,YANG Z L,DU P,et al. Effects of lithium sources on the performance of LiNi0.5Co0.2Mn0.3O2cathode material derived by solid-state method[J]. Journal of South China Normal University (Natural Science Edition),2015,47(2):48-52.
[12] 谭春林,周豪杰,许梦清,等. 铋修饰LiMn2O4锂离子电池正极材料的研究[J]. 华南师范大学学报(自然科学版),2013,45(4):100-103.
TAN C L,ZHOU H J,XU M Q,et al. Study onbismuth modified LiMn2O4as cathode of lithium ion battery[J]. Journal of South China Normal University (Natural Science Edition),2013,45(4):100-103.
[13] KOLLMAN P A,LIEBMAN J F,ALLEN L C. Thelithium bond[J]. Journal of the American Chemical Society,1970,92(5):1142-1150.
[14] AULT B S,PIMENTEL G C. Matrixisolation infrared stu-dies of lithium bonding[J]. Journal of Physical Chemistry,1975,79(6):621-626.
[15] FENG Y,LIU L,WANG J T,et al. Blue-shifted lithium bonds[J]. Chemical Communications,2004,1(1):88-89.
[16] SHAHI A,ARUNAN E. Hydrogen bonding,halogen bonding and lithium bonding:an atoms in molecules and natural bond orbital perspective towards conservation of total bond order,inter-and intra-molecular bonding[J]. Physical Chemistry Chemical Physics,2014,16(42):22935-22952.
[17] DUNNING T H,Gaussianbasis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen [J]. Journal of Chemical Physics,1989,90 (2):1007-1023.
[18] PRASCHER B P,WOON D E,PETERSON K A,et al. Gaussianbasis sets for use in correlated molecular calculations. VII. Valence,core-valence,and scalar relativistic basis Sets for Li,Be,Na,and Mg[J]. Theoretical Chemistry Accounts. 2011,128(1):69-82.
[19] CHAI J D,HEAD-GORDON M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections[J]. Physical Chemistry Chemical Phy-sics,2008,10(44):6615-6620.
[20] WIBERG K B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane[J]. Tetrahedron,1968,24(3):1083-1096.
[21] FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al. Gaussian 09,Revision A.01 [EB/OL]. http://www.gaussian.com/citation_g09.htm.
[22] STANTON J F,GAUSS J,WATTS J D,CFOUR,version 1.0 [EB/OL]. http://www.cfour.de.
[23] PIPEK J,MEZEY P G. A fast intrinsic localization procedure applicable for abinitio and semiempirical linear combination of atomic orbital wave functions[J]. Journal of Chemical Physics,1989,90(9):4916-4926.
[24] HUBER K P,HERZBERG G. Molecular spectra and molecular structure,constants of diatomic molecules[M]. vol. 4. New York:Van Nostrand Reinhold,1979.
[25] LOVAS F J,TIEMANN E. Microwavespectral tables I. diatomic molecules[J]. Journal of Physical and Chemical Reference Data,1974,3(3):609-770.
[26] JOHNSON M W,SANDOR E,ARZI E. Thecrystal structure of deuterium fluoride[J]. Journal of Physical and Chemical Reference Data,1975,31(8):1998-2003.
Theoretical Studies on Hydrogen Bond and Lithium Bond in X…Y(X=LiF,NH3,H2O; Y=HF,LiF) Complexes
LIU Yameng1, LI Jun2, GAO Aifang3, ZHEN Yan4*
(1. College of Life Science & Bioengineering, Beijing University of Technology, Beijing Key Laboratory of Environmental and Viral Oncology, Beijing 100124,China; 2. Department of Chemistry, Guangdong University of Education, Guangzhou 510303,China; 3. School of Water Resources and Environment, Hebei GEO University, Shijiazhuang 050031,China;4. College of Environmental and Energy Engineering, Beijing University of Technology, Beijing 100124,China)
Nine structures of X…Y(X=LiF,NH3,H2O; Y=HF,LiF) complexes were optimized at CCSD(T)/cc-pVTZ level. The results of natural atomic charges, Wiberg bond index and localized molecular orbitals suggest that H—F bond in HFLiF is covalent, but Li—F bond is more like ionic instead of covalent. The bind energy of LiF with X (X=LiF,H2O,NH3) is higher than that of X…HF. The order of bind energy is LiF…Y> H3N…Y > H2O…Y (Y=LiF,HF). The obtained vibrational frequencies of H—F bond in H3N…HF or H2O…HF decrease while H—F bond length increase, suggesting a red-shifting hydrogen bond. The red-shifting lithium bond is also found in H3N…LiF. In H2O…LiF, it is found that Li—F bond elongated 0.001 6 nm while the stretch vibrational frequency of Li—F increased 2 cm-1. Thus, the lithium bond in H2O…LiF should be considered as blue-shifting lithium bond.
2016-12-31 《华南师范大学学报(自然科学版)》网址:http://journal.scnu.edu.cn/n
广东省高层次人才项目(9010-14193);河北省青年拔尖人才计划
*通讯作者:甄岩,教授,Email:zhenyan@bjut.edu.cn.
O43
A
1000-5463(2017)05-0043-05
【中文责编:谭春林 英文审校:李海航】
猜你喜欢
大学物理(2022年9期)2022-09-28
高中数理化(2022年14期)2022-08-15
波谱学杂志(2021年3期)2021-09-07
物理通报(2020年7期)2020-07-01
青岛大学学报(工程技术版)(2019年2期)2019-09-10
信阳师范学院学报(自然科学版)(2018年1期)2018-08-09
中学化学(2015年12期)2016-01-19
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
原子与分子物理学报(2015年3期)2015-11-24
