
亚洲棉EST-SNP的挖掘及其在陆地棉中的验证
2017-10-19 05:57徐鹏蔡继鸿郭琪张香桂徐珍珍沈新莲
棉花学报 2017年5期
徐鹏,蔡继鸿,郭琪,张香桂,徐珍珍,沈新莲
亚洲棉EST-SNP的挖掘及其在陆地棉中的验证
徐鹏,蔡继鸿,郭琪,张香桂,徐珍珍,沈新莲*
(江苏省农业科学院经济作物研究所/农业部长江下游棉花油菜重点实验室,江苏南京210014)
【目的】随着不同棉种序列数据库的逐步完善以及高通量测序技术的发展,棉花单核苷酸多态性(Single nucleotidepolymorphism,SNP)标记开发可利用的公共数据资源逐步增加。【方法】本研究基于陆地棉祖先基因组的现代种亚洲棉表达序列标签(Expressed sequencetag,EST)数据库,利用CAP3对亚洲棉EST数据库进行拼接。拼接获得7 187个重叠群(Contig),再利用QualitySNP软件进行SNP位点分析。【结果】在807条含有4条以上EST序列的Contig中查找到2 690个SNP位点。通过筛选次要等位基因频率大于30%的位点,获得953个可靠度较高的候选SNP,通过电子筛选,最终获得可用于陆地棉分析的SNP 149个,利用位点特异性聚合酶链式反应以及酶切扩增多态序列验证了EST-SNP的准确性。【结论】本研究证实基于亚洲棉EST数据库挖掘用于陆地棉研究的EST-SNP切实可行,并有望将EST-SNP用于陆地棉遗传图谱构建、重要性状的基因定位以及分子标记辅助育种。
亚洲棉;单核苷酸多态性;酶切扩增多态性序列;表达序列标签
Abstract:[Objective]With the development of the cotton genome sequence database and next-generation high-throughput sequencing techniques,the resources available for generating single nucleotide polymorphism (SNP)markers are gradually expanding.[Method]The Gossypium arboreum expressed sequence tags(ESTs)downloaded from the NCBIdatabase were assembled into 7 187 contigs using the CAP3 program.Additionally,the QualitySNPprogram was used for SNPmining.[Result]A total of 2 690 SNPs were obtained from 807 contigs that consisted of more than four ESTs.We obtained 953 highly reliable candidate SNPsby screening for a minor loci frequency of more than 30%.Finally,a total of 149 candidate EST-SNPsthat may be used in G.hirsutum were obtained through in silico screening.An allele-specific polymerase chain reaction and cleaved amplified polymorphic sequencemolecular markerswereused to validatethe accuracy of the selected candidate SNPsin G.hirsutum.[Conclusion]The EST-SNPmarkers from G.arboreum may be used to analyze G.hirsutum.The obtained EST-SNPmarkers will beused to construct genetic maps,map important traits,and completemarker-assisted selection in G.hirsutum.
Keywords:Gossypium arboreum;single nucleotide polymorphism;cleaved amplified polymorphic sequence;expressed sequence tag
目前棉花分子标记的开发和应用主要集中于利用公用数据库开发的简单重复序列(Simple sequence repeat,SSR)分子标记。由于陆地棉的遗传基础狭窄,种内遗传图谱所含的SSR标记数较少,覆盖率低,不能满足实际育种的需要;所以,开发更多其他类型的分子标记成为研究者迫切需要解决的问题。单核苷酸多态性 (Single nucleotide polymorphism,SNP)分子标记广泛分布于基因组范围内,具有变异来源丰富、潜在数量巨大等优点,具有广阔的应用前景[1]。
较高的前期测序成本是大规模开发SNP标记的主要限制因素。因此,通过生物信息学方法利用现有数据进行SNP位点的查找并通过后期试验加以验证已成为1种快捷高效的SNP开发途径。随着不同棉种序列数据库的逐步完善以及高通量测序技术的发展,棉花SNP标记开发的可利用公共数据资源逐步增加,使得利用表达序列标签(Expressed sequence tag,EST)进行 SNP 标记的开发具有很多的优势。当前,异源四倍体棉花中存在2个不同进化来源的亚基因组,在进行SNP检测时,亚基因组间和亚基因组内SNP位点的区分较困难。通过序列比对所发现的SNP很多是部分同源(异源多倍体内亚基因组间的同源性)序列间的差异,而不是等位同源序列间的差异[2]。陆地棉A、D亚基因组种间同源序列的差异使得基于陆地棉EST开发SNP具有较高的假阳性,开发效率很低。亚洲棉(Gossypium arboreum)、雷蒙德氏棉(G.raimondii)被认为是异源多倍体祖先基因组的现代种。陆地棉中具有部分同源性的A、D亚基因组分别与亚洲棉、雷蒙德氏棉的基因组具有极小的序列分歧[3]。二倍体与多倍体亚基因组间的相似性、共线性为多倍体陆地棉基因组中区分SNP提供了材料基础。
SNP标记检测方法由于操作复杂、成本昂贵,需要高端的仪器设备;因此,SNP标记技术在动植物遗传育种中的应用受到了严重限制。酶切扩增多态性序列 (Cleaved amplified polymorphic sequence,CAPS)是1种将特异引物聚合酶链式反应(Polymerasechain reaction,PCR)与限制性内切酶消化结合的分子标记检测技术,具有共显性、位点特异性、操作简单和成本低等特点,是检测SNP位点的常用方法[4]。本研究基于陆地棉祖先基因组的现代种亚洲棉EST数据库,利用生物信息学手段大规模查找SNP位点,通过电子筛选,获得可用于陆地棉分析的SNP,对部分SNP位点在陆地棉中进行位点特异性PCR以及对含有酶切位点的SNP位点在陆地棉中CAPS验证,以期用于陆地棉遗传图谱构建、重要性状的基因定位以及分子标记辅助育种。
1 材料与方法
1.1 供试材料
截至2016年6月,61 898条亚洲棉EST序列下载于美国国立生物技术信息中心 (NCBI)网站(http://www.ncbi.nlm.nih.gov/),所有 EST 序列以fasta格式保存。
用于SNP验证的陆地棉品种分别为DP555、Miscott7913-83、苏 12、枝棉 3 号、徐州 142、鲁棉研28。
1.2 EST序列的聚类和拼接
为了得到更可靠的SNP位点,首先要对EST序列进行预处理,从而获得clean序列用于后续SNP位点分析。利用Cross_match[5]对EST序列进行载体序列屏蔽,通过 EST_trimmer.pl(http://pgrc.ipk-gatersleben.de/misa/download/est_trinimer.pl)脚本进行PolyA以及过短和过长EST的处理, 具体参数为-amb=2,50;-tr5=T,5,50;-tr3=A,5,50;-cut=100 700(参数含义分别为去掉末端50 bp内不明确的碱基,去掉5'的Poly T,去掉3'端Poly A,选择长度介于100到700 bp的序列);CAP3软件对EST序列进行聚类与拼接,拼接最小重叠碱基为100 bp,具体参数为-p 95-o 100(相似度95%,最小重叠碱基数100 bp)。
1.3 EST-SNP位点的挖掘
提取在CAP3拼接结果中4条及以上的EST序列重叠群用于SNP位点开发。利用软件Qual itySNP[6](http://www.bioinformatics.nl/tools/snpweb/)对含有4条以上亚洲棉EST序列的重叠群开发SNP位点,同时分析SNP的类型,并对次要等位基因的频率进行筛选。
1.4 候选SNP位点的验证
根据突变位点设计特异PCR引物(http://ausubellab.mgh.harvard.edu/,SNAP program)。 引物设计的主要参数:引物长度20~36 bp;Tm62~70℃(最适为67℃);GC含量为45%~65%,最适为 50%。95℃预变性 5 min;94℃变性30 s,65℃退火45 s,72℃延伸 1 min,36个循环;72℃延伸 10 min;4 ℃保存。产物在 12 g·L-1琼脂糖凝胶上电泳,观察电泳条带是否具有多态性。
在候选SNP中随机选取具有酶切位点的候选SNP进行CAPS验证。首先保证目标序列中只存在1个目标酶切位点,然后在目标酶切位点两侧用Primer5.0进行引物设计,引物设计的主要参数为引物长度18~22 bp(最适为20 bp),Tm55~65℃(最适为60℃),GC含量为45%~65%(最适为50%)。95℃预变性5 min;94℃变性30 s,60 ℃退火 45 s,72 ℃延伸 1 min,36 个循环;72℃延伸10 min;4℃永久保存。酶切分析参照TaKaRa的内切酶操作指南,用限制性核酸内切酶酶切PCR产物,酶切体系包括10 U·μL-1限制酶 0.3μL、1μL buffer、5.7μL ddH2O、3μL PCR 产物,酶切2 h。酶切完毕后酶切产物在12 g·L-1琼脂糖凝胶上电泳,观察电泳条带的多态性。
2 结果与分析
2.1 亚洲棉EST的处理及拼接
利用Cross_match和EST_trimmer.pl对原始EST序列进行预处理后,最终获得57 308条clean亚洲棉EST序列。利用CAP3对clean EST拼接,拼接最小重叠碱基为100 bp,序列相似度为95%。亚洲棉EST拼接后获得7 187条重叠群(Contig),平均每个重叠群含有4.54条EST,含有2条EST序列的重叠群3 363条,含有3条EST序列的重叠群1 336条,含有4条及以上EST序列的重叠群2 488条,24 688条EST未参与拼接(表 1)。

表1 亚洲棉EST序列拼接结果Table 1 Assembling results of G.arboreum EST
2.2 EST-SNP位点分析
由于序列的拼接需要大量的冗余序列作为基础,才能保证候选SNP的可靠度,所以本研究选择2 488条含有4条以上亚洲棉EST序列的Contig利用QualitySNP软件查找SNP位点 (图1)。结果表明,所有用于开发SNP的Contig序列总长为2 293 541 bp,其中807个Contig含2 690个候选SNP,平均每852 bp出现 1个SNP,每个Contig含有1~87个SNP,平均每个Contig含有3.33个SNP。随着Contig所含的EST数目的增加,SNP数目也表现为逐渐增加的趋势(图2)。其中Contig88由132条EST拼接而成,共含有87个SNP位点。在2 690个候选SNP位点中,颠换类型有1 139个,转换类型1 106个,单碱基插入缺失类型445个。其中A-G转换类型的SNP最多,占22.71%;其次为C-T转换类型,占18.40%;C-插入缺失类型则最少,仅占2.83%(表2)。为了提高候选SNP的可靠度,进一步筛选候选SNP次要等位基因的频率至少为30%的SNP位点,最终获得953个可靠度较高的候选SNP用于后续的分析。
2.3 可用于陆地棉分析的EST-SNP的电子筛选及其验证

图1 EST-SNP位点的挖掘Fig.1 Identification of EST-SNP
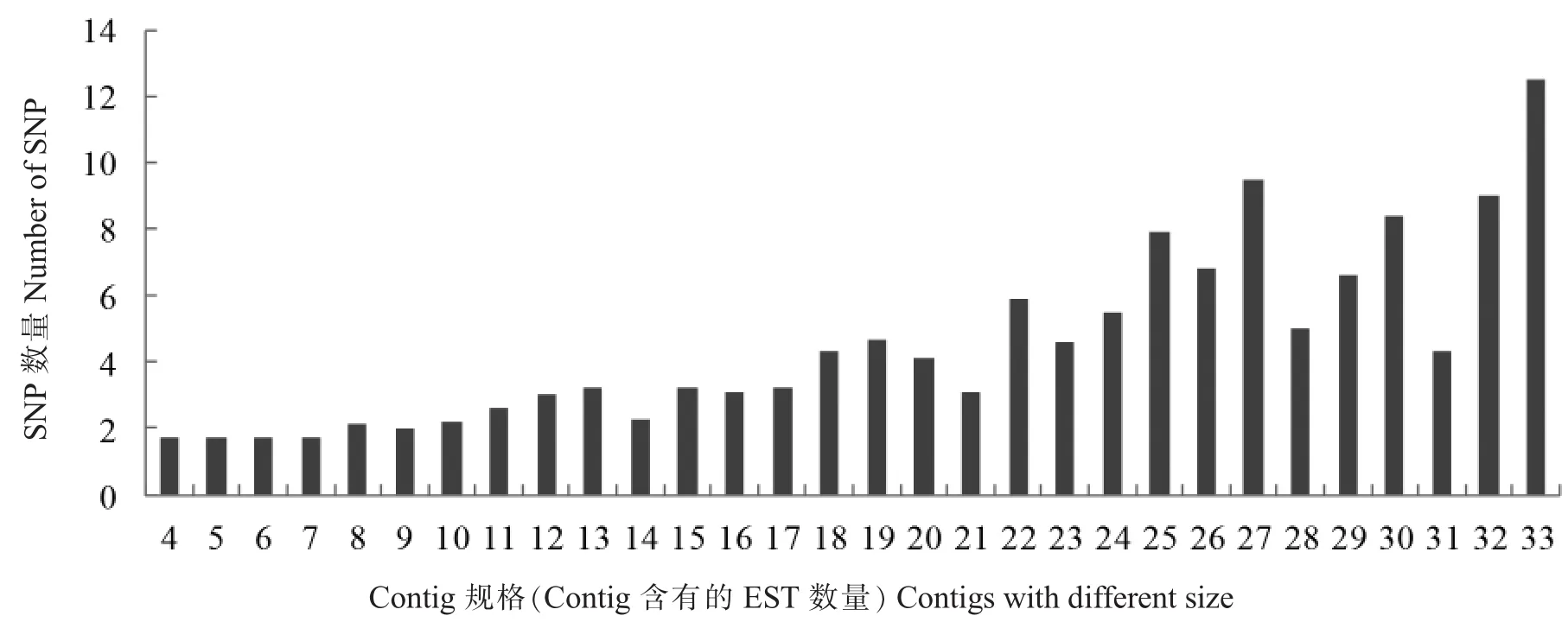
图2 不同规格重叠群平均候选SNP数量Fig.2 Average amounts of candidate SNP from different contigs

表2 候选SNP的类型分析Table 2 Analysis of candidate SNP types
利用以上获得的807条含候选SNP的Contig作为查询序列,以陆地棉EST数据库(下载于http://www.ncbi.nlm.nih.gov/)作为参考序列进行本地 Blastn[7](E-value<10-10),获得与 807 条亚洲棉Contig联配的陆地棉EST 40 728条。以QualitySNP分析后产生的allavailsnp文件作为查询序列,以上获得的40 728条与亚洲棉Contig联配的陆地棉EST作为参考数据库,利用短序列比对软件 bowtie(http://bowtie-bio.sourceforge.net/index.shtml)进行比对,总共681个SNP能够比对到参考数据库(图3),其中532个SNP位点只有1个基因型能够匹配,即该类型的SNP在陆地棉中表现为单态性。筛选SNP位点2种基因型均能够与陆地棉EST完全匹配的序列作为候选,最终获得149个可用于陆地棉分析的候选EST-SNP(表 3)。
为了验证候选SNP的可靠性,从149个候选的EST-SNP中随机选择位点进行验证。为了保证获得合适大小的PCR产物,随机选择SNP位于序列5'端的Contig2325,利用SNAPprogram根据突变位点设计特异PCR引物Contig2325-F:5'-AATGGCTTCCATGCTTAGCTCTGGACT-3',Contig2325-R:5'-CAAAGGCCTCAGGGTCGGCTG-3'。验证结果表明,在不同的陆地棉品种中Contig2325的候选SNP具有多态性(图4)。

图3 候选SNP在陆地棉EST中的匹配分析Fig.3 Analysis of SNP mapping to EST database of Gossypium hirsutum
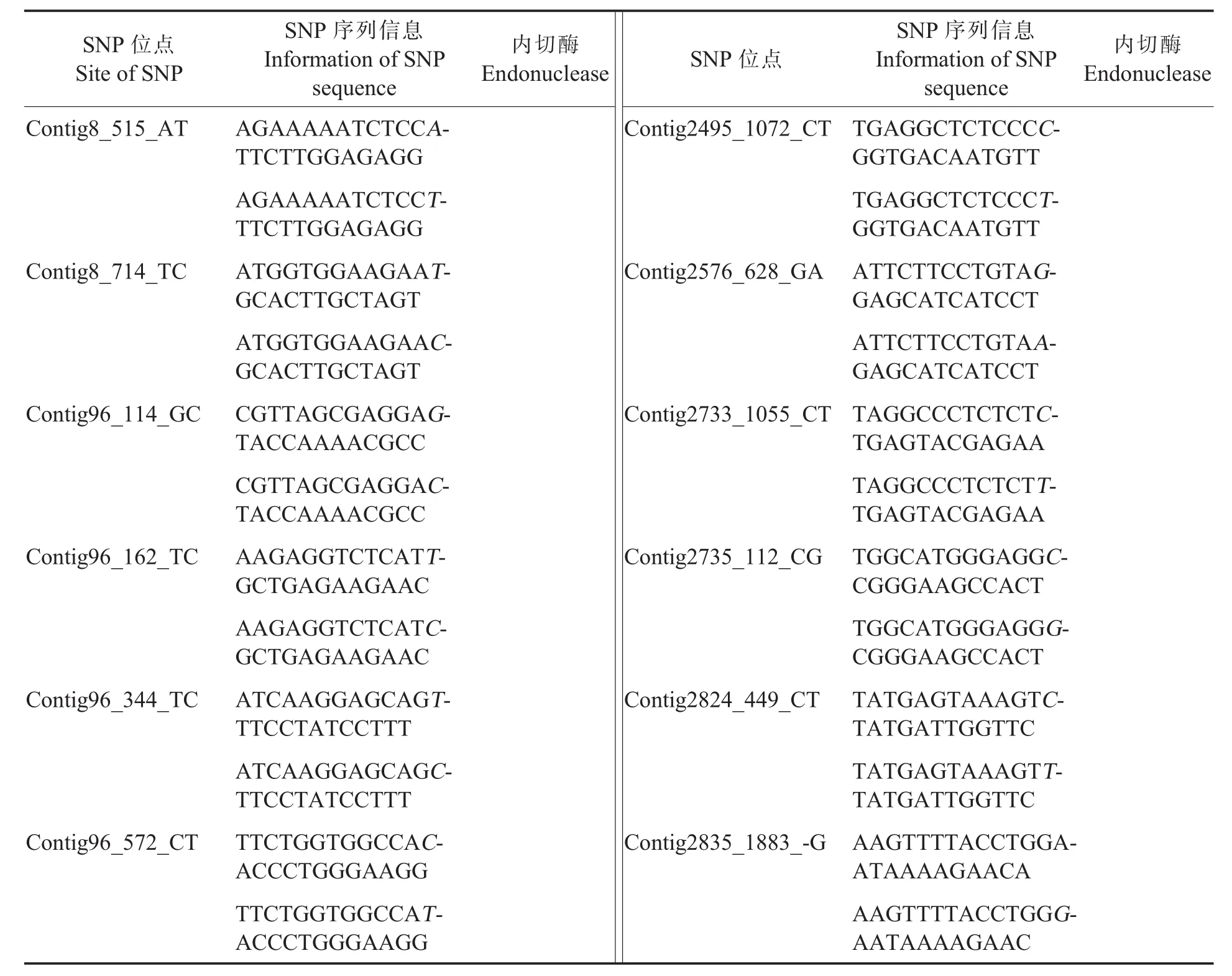
表3 149个可用于陆地棉分析的候选EST-SNP信息Table 3 Information of 149 candidate EST-SNPs
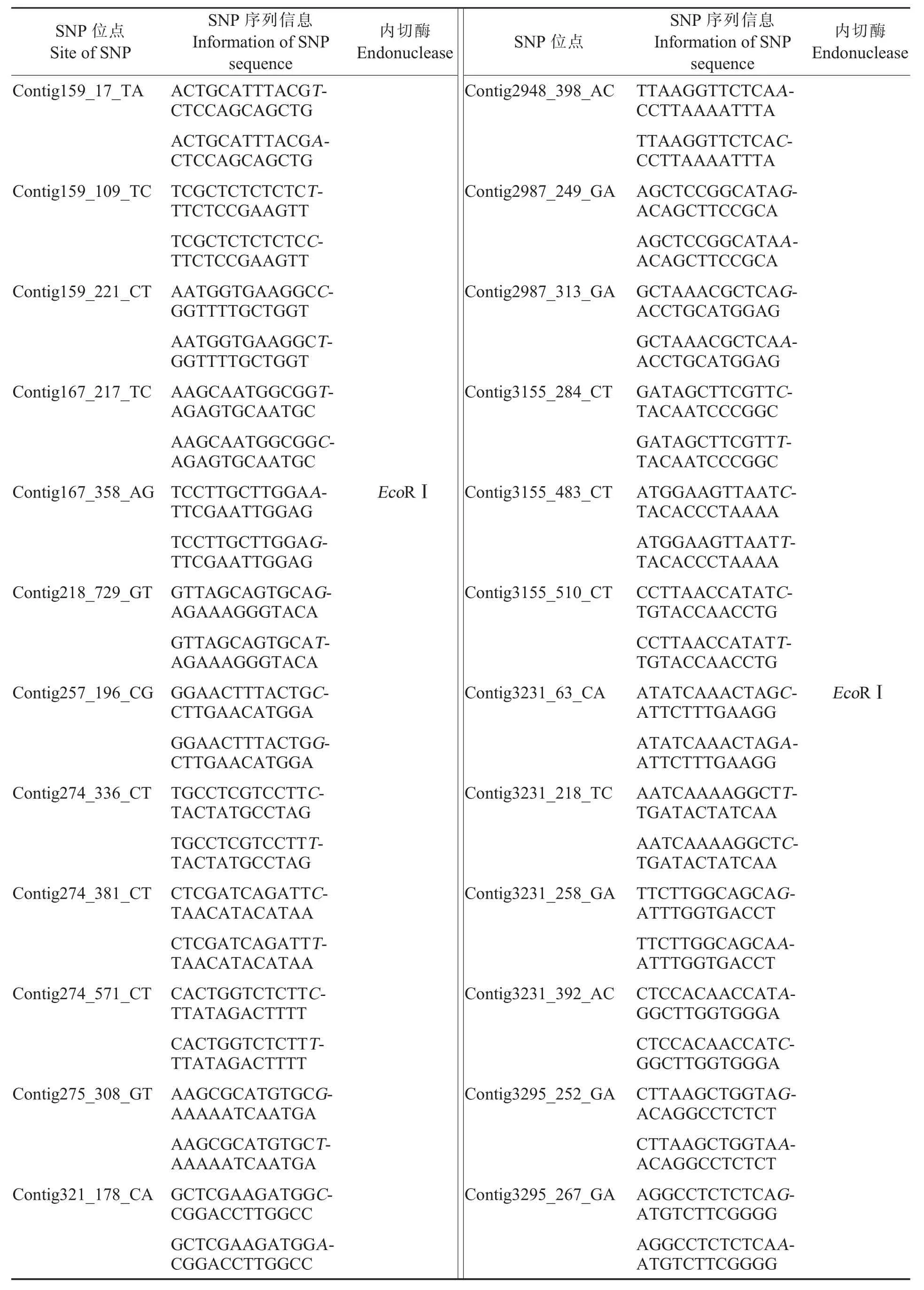
表3 (续)Table 3(Continued)
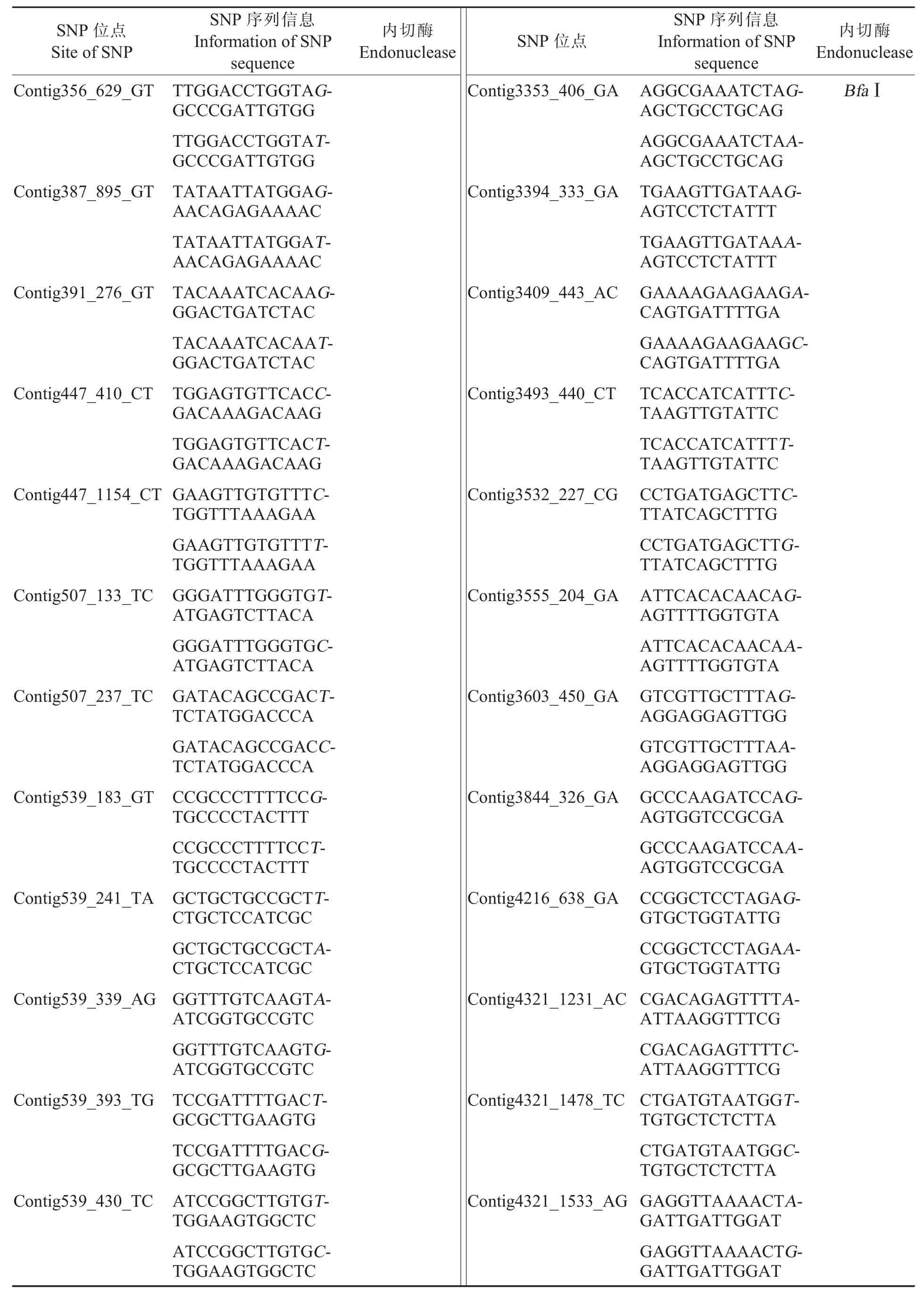
表3 (续)Table 3(Continued)
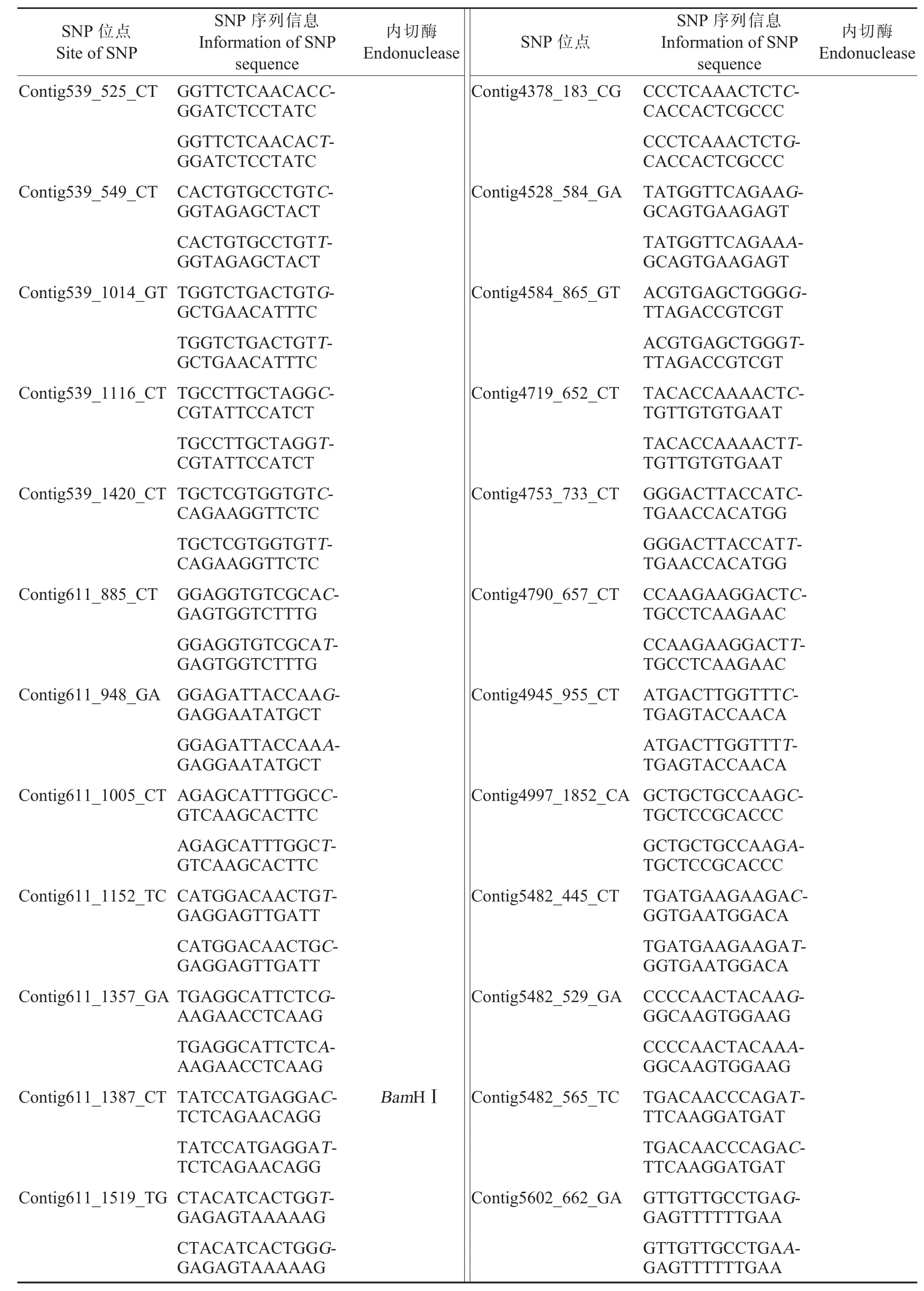
表3 (续)Table 3(Continued)
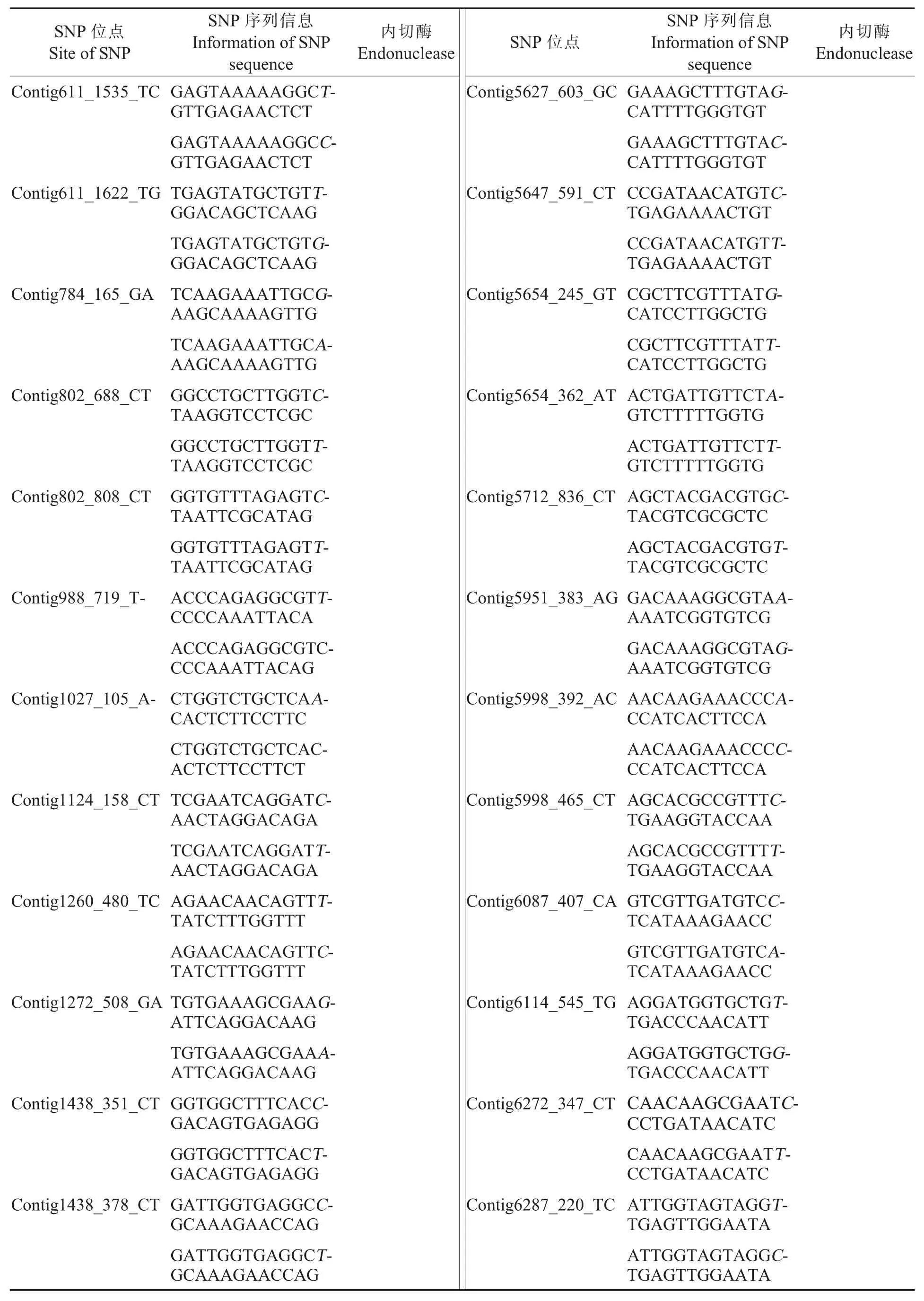
表3 (续)Table 3(Continued)
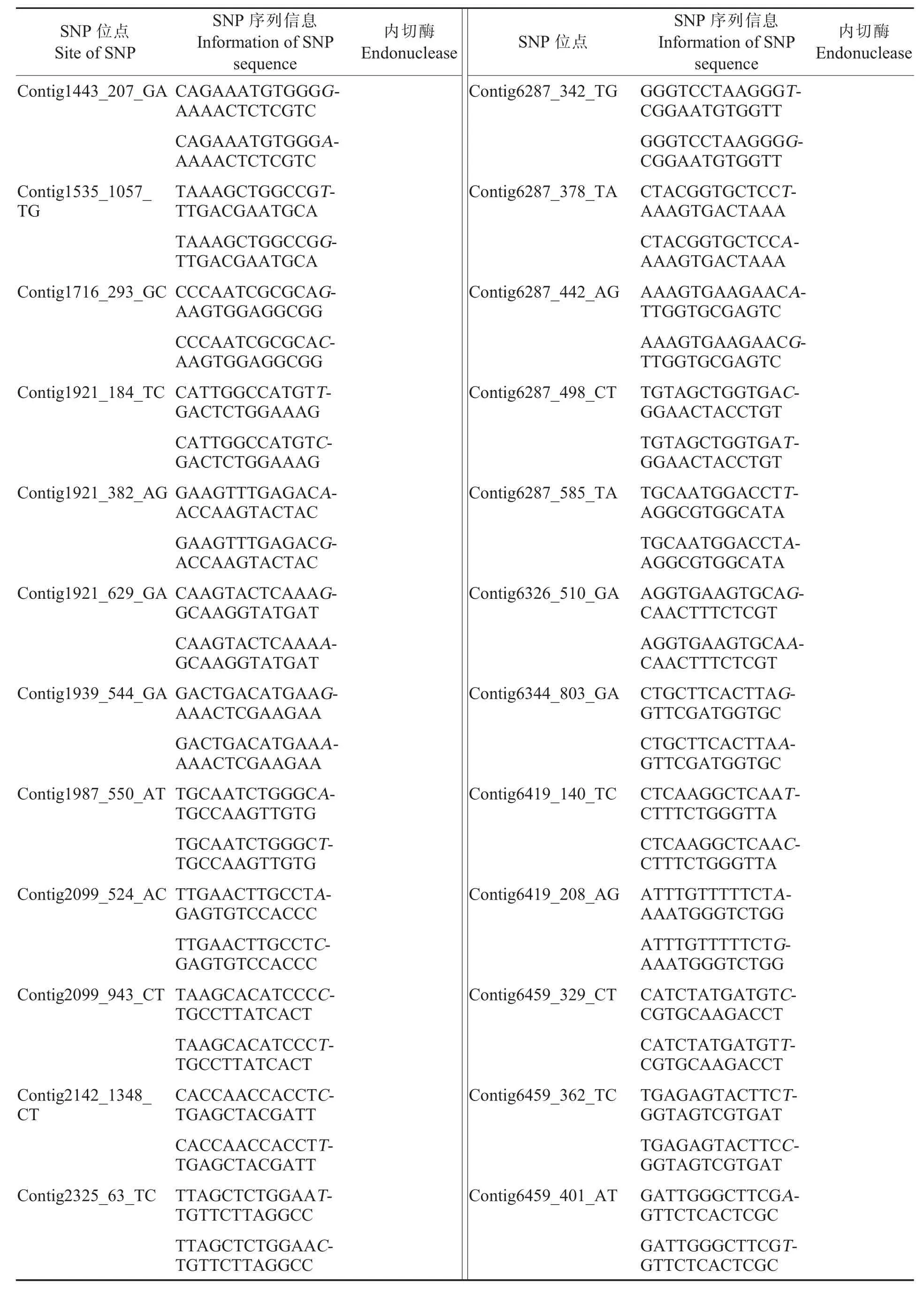
表3 (续)Table 3(Continued)

表3 (续)Table 3(Continued)
随机选取具有酶切位点的候选SNP进行CAPS验证,用酶切位点识别序列去搜索149条候选的SNP序列信息。本研究用常用的限制性内切酶识别序列去搜索allavailsnp文件中149个SNP序列信息 (表 3), 发现 Contig167、Contig3231在 Eco RⅠ酶切位点处有 SNP。由于Contig3231的酶切位点识别序列位于序列的5'端,无法设计引物;因此,仅对Contig167设计引物,引物序列为Contig167-F:5'-CATACCTCCC CGATCTTACACC-3',Contig167-R:5'-ACTAATGCACTGCACTTGACGC-3'。PCR扩增酶切产物的电泳结果显示,Contig167的候选SNP具有多态性(图5)。因此,通过亚洲棉EST开发用于陆地棉分析的EST-SNP基本可行。
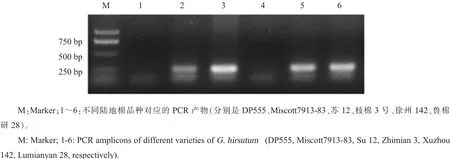
图4 Contig2325候选SNP位点特异性PCR验证Fig.4 Validation of the candidate SNP of the contig2325 using allele-specific PCR
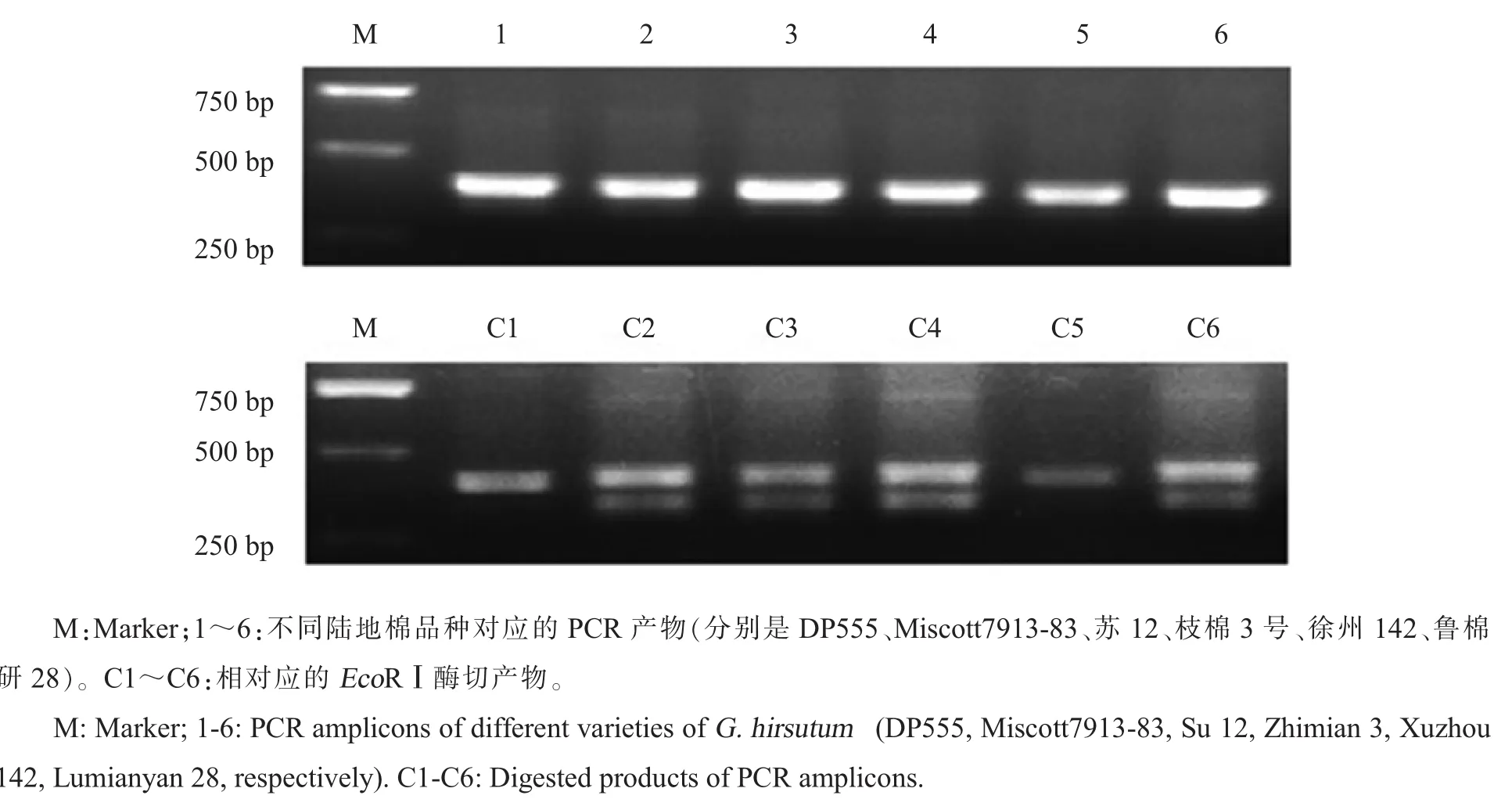
图5 Contig167候选SNP位点的CAPS验证结果Fig.5 Validation of the candidate SNP of the contig167 using CAPS
3 讨论
利用现有数据,结合生物信息学知识及相关分析软件进行SNP标记开发,再制定针对候选SNP位点的验证方法,因其具有开发成本低快捷高效等优点,而被广大科研工作者青睐。公共数据库中已经积累了大量的EST序列,很多来源于不同的个体或品种,大量的冗余序列拼接时往往会出现不一致的碱基,即为EST-SNP突变位点,这些位点可以通过生物信息学方法检测到[8]。近些年来,在植物方面从模式植物拟南芥[9],到主要粮食作物水稻[10]、玉米[11]、小麦[12]和大麦[13],及一些小物种植物,如番茄[14]、松树[15]、苹果[16]等,该方法均得到了普遍应用。EST来源于功能基因表达的cDNA片段,相关公共数据库中增速最快的核苷酸序列是EST序列,使得以EST序列为基础进行相关分子标记开发变得越来越方便。同时利用EST序列开发出的候选SNP位点很可能与表达基因紧密相关或直接位于基因的编码区内,可直接应用于植物分子育种的研究实践。本研究基于陆地棉祖先基因组的现代种亚洲棉EST数据库,利用软件QualitySNP查找到953个可靠度较高的SNP位点,通过在陆地棉EST数据库中电子筛选,最终获得149个可用于陆地棉分析的候选EST-SNP,以期用于陆地棉遗传图谱构建、重要性状的基因定位以及分子标记辅助育种的研究。
棉花上SNP标记大规模开发的首次报道是在2009年Van Deynze等[17]以陆地棉和海岛棉为主要研究对象,开发了约1 000个海陆棉种之间的SNP。随着测序技术的发展,在棉花SNP标记的开发方面已获得初步进展。Hulse-Kemp等[18]首先对陆地棉遗传标准系TM-1的细菌人工染色体(Bacterial artificial chromosome,BAC) 文库进行末端测序,然后利用这些末端序列为参考对12个陆地棉材料、1个海岛棉及1个长萼棉(G.longicalyx Hutchinson&Lee)基因组重测序并进行序列比对,在12个陆地棉材料中发现了132 262个种内SNP标记,在陆地棉与海岛棉间挖掘到了223 138个SNP标记,在陆地棉与长萼棉间挖掘了70631个SNP标记。Zhu等[19]通过简化基因组测序 (RAD-seq)在22个陆地棉品种中得到3 090个SNP标记。目前利用公共数据库开发棉花SNP的研究则报道较少。Li等[20]利用HaploSNPer软件对收集到的陆地棉和海岛棉的EST进行序列比对,开发出了356个SNP标记。
棉花栽培种主要为异源四倍体,A和D亚基因组间的部分同源序列区分困难,难以区别棉花中2个亚基因组间的单核苷酸变异和亚基因组内的单核苷酸变异。这在很大程度上阻碍了棉花中SNP标记的开发和应用进程。此外,棉花测序工作完成较晚,无法提供参考基因组,这些对棉花中SNP标记的开发进程有一定的影响。陆地棉中具有部分同源性的A、D亚基因组分别与亚洲棉、雷蒙德氏棉的基因组具有极小的序列分歧[3]。随着不同棉种基因组测序的完成,基于基因组重测序能够快速找到大量的基因组变异,因此它是目前发掘SNP标记最强大的工具。Wang等[21]对陆地棉TM-1和海岛棉海7124进行了基因组重测序,以TM-1基因组序列作为参考序列,共在2个材料间鉴定出6 476 899个SNP标记。
4 结论
本研究基于陆地棉祖先基因组的现代种亚洲棉EST数据库,消除部分同源序列之间的干扰,提高了开发效率,证实了通过亚洲棉EST开发用于陆地棉基因组分析的EST-SNP的可行性。
[1]Ganal M W,Altmann T,Röder M S.SNPidentification in crop plants[J].Current Opinionin Plant Biology,2009,12(2):211-217.
[2]Kaur S,Francki M G,Forster JW.Identification,characterization and interpretation of single-nucleotide sequence variation in allopolyploid crop species[J].Plant Biotechnology Journal,2012,10(2):125-138.
[3]Senchina D S,Alvarez I,Cronn R C,et al.Rate variation among nuclear genesand theageof polyploidy in Gossypium[J].Molecular Biology and Evolution,2003,20(4):633-643.
[4]Lee GA,Koh H J,Chung H K,et al.Development of SNP-based CAPS and dCAPS markers in eight different genes involved in starch biosynthesis in rice[J].Molecular Breeding,2009,24(1):93-101.
[5]Ewing B,Green P.Base-calling of automated sequencer traces using phred.Ⅱ.Error probabilities[J].Genome Research,1998,8:186-194.
[6]Tang Jifeng,Vosman B,Voorrips R E,et al.QualitySNP:a pipeline for detecting single nucleotide polymorphisms and insertions/deletionsin EST datafrom diploid and polyploid species[J/OL].BMCBioinformatics,2006,7:438[2016-10-17].http://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-7-438.DOI:10.1186/1471-2105-7-438.
[7]Altschul S F,Madden T L,Schaffera A,et al.Gapped BLAST and PSI-BLAST:A new generation of protein database search program[J].Nucleic Acids Research,1997,25:3389-3402.
[8]Picoultnewberg L,Ideker T E,Pohl M G,et al.Mining SNPs from EST database[J].Genome Research,1999,9(2):167-174.
[9]Torjk O,Berger D,Meyer RC,et al.Establishment of a high-efficiency SNP-based framework marker set for Arabidopsis[J].The Plant Journal,2003,36(1):122-140.
[10]Feltus FA,Wan J,Schulze SR,et al.An SNPresource for rice genetics and breeding based on subspecies indica and japonica genome alignments[J].Genome Research,2004,14(9):1812-1819.
[11]Batley J,Barker G,O'Sullivan H,et al.Mining for single nucleotide polymorphisms and insertions/deletions in maize expressed sequence tag data[J].Plant Physiology,2003,132(1):84-91.
[12]Rustgi S,Bandopadhyay R,Balyan H S,et al.EST-SNPs in bread wheat:discovery,validation,genotyping and haplotype structure[J].Czech Journal of Geneticsand Plant Breeding,2009,45:106-116.
[13]Kota R,Varshney RK,Prasad M,et al.EST-derived single nucleotide polymorphism markers for assembling genetic andphysical mapsof thebarley genome[J].Functional&Integrative Genomics,2008,8(3):223-233.
[14]Yamamoto N,Tsugane T,Watanabe M,et al.Expressed sequence tags from the laboratory-grown miniature tomato(Lycopersicon esculentum)cultivar Micro-Tom and mining for single nucleotide polymorphisms and insertions/deletions in tomato cultivars[J].Gene,2005,356:127-134.
[15]Dantec L L,ChagnéD,Pot D,et al.Automated SNPdetection in expressed sequence tags:Statistical considerations and application to maritime pine sequences[J].Plant Molecular Biology,2004,54(3):461-470.
[16]ChagnéD,Gasic K,Crowhurst RN,et al.Development of a set of SNPmarkers present in expressed genes of the apple[J/OL].Genomics,2008,92(5):353[2016-10-17].http://www.sciencedirect.com/science/article/pii/S0888754308001808.DOI:10.1016/j.ygeno.2008.07.008.
[17]Van Deynze A,Stoffel K,Lee M,et al.Sampling nucleotide diversity in cotton[J/OL].BMC Plant Biology,2009,9(1):125[2016-10-17].https://bmcplantbiol.biomedcentral.com/articles/10.1186/1471-2229-9-125.DOI:10.1186/1471-2229-9-125.
[18]Hulse-Kemp A M,Ashrafi H,Stoffel K,et al.BAC-end sequence-based SNPmining in allotetraploid cotton(Gossypium)utilizing resequencing data,phylogenetic inferences,and perspectives for genetic mapping[J].Genes Genomes Genetics,2015,5(6):1095-1105.
[19]Zhu Qianhao,Spriggs A,Taylor JM,et al.Transcriptome and complexity-reduced,DNA-based identification of intraspecies single-nucleotide polymorphisms in the polyploid Gossypium hirsutum L.[J].Genes Genomes Genetics,2014,4(10):1893-1905.
[20]Li Ximei,Gao Wenhui,Guo Huanle,et al.Development of EST-based SNP and InDel markers and their utilization in tetraploid cotton genetic mapping[J/OL].BMC Genomics,2014,15(1):1046[2016-10-17].http://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-15-1046.DOI:10.1186/1471-2164-15-1046.
[21]Wang Sen,Chen Jiedan,Zhang Wenpan,et al.Sequence-based ultra-dense genetic and physical maps reveal structural variations of allopolyploid cotton genomes[J/OL].Genome Biology,2015,16(1):108[2016-10-17].http://genomebiology.biomedcentral.com/articles/10.1186/s13059-015-0678-1.DOI:10.1186/s13059-015-0678-1. ●
Development of EST-SNP Markers in Gossypium arboreum and Their Validation in G.hirsutum
Xu Peng,Cai Jihong,Guo Qi,Zhang Xianggui,Xu Zhenzhen,Shen Xinlian*
(Institute of Industrial Crops,Jiangsu Academy of Agricultural Sciences/Key Laboratory of Cotton and Rapeseed,Ministry of A-griculture,Nanjing 210014,China)
S562.03
A
1002-7807(2017)05-0401-14
10.11963/1002-7807.xpsxl.20170628
2016-10-17 第一作者简介:徐鹏(1981―),男,Semon528@hotmail.com。*通信作者:xlshen68@126.com
国家自然科学基金(31471545);江苏省自然科学基金(BK20160580);国家科技重大专项——转基因生物新品种培育(2014ZX08005-004-002);棉花生物学国家重点实验室开放课题(CB2015A12);江苏省协同创新中心
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
昆明医科大学学报(2022年2期)2022-03-29
上海金属(2021年6期)2021-12-02
今日农业(2021年11期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
烟草科技(2021年6期)2021-06-24
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
电脑知识与技术(2018年19期)2018-11-01
中成药(2018年7期)2018-08-04
