
M(H2O)1~4 ( M=Li+,Na+,Be2+和Mg2+)的分子形貌理论研究
2017-09-22 09:43:40赵东霞朱尊伟杨忠志
辽宁师范大学学报(自然科学版) 2017年3期
赵东霞, 朱尊伟, 杨忠志
(辽宁师范大学 化学化工学院, 辽宁 大连 116029)
M(H2O)1~4( M=Li+,Na+,Be2+和Mg2+)的分子形貌理论研究
赵东霞, 朱尊伟, 杨忠志
(辽宁师范大学 化学化工学院, 辽宁 大连 116029)
金属离子与水分子的相互作用机理,对理解含金属离子生物分子的结构和功能具有指导意义.以M(H2O)1~4( M = Li+,Na+,Be2+和Mg2+)为模型分子,基于分子中单电子受到的作用势,绘制了各个体系的分子形貌.结果表明:在内禀特征轮廓上,电子密度最大值出现在氧原子附近,而最小值出现在金属离子附近.计算了各个体系金属离子和氧原子之间,化学键键心处的PAEM值以及电子密度临界点的电子密度.为了分析水分子与金属离子结合的强弱程度,计算了各体系的连续结合能.结果显示,键心处PAEM值和连续结合能具有线性关系.对讨论金属离子与其他分子之间的相互作用,有参考价值.
单电子作用势;分子形貌;Dpb;连续结合能;水合金属离子
金属及其离子在自然界中广泛分布,在化学和生物化学中扮演着重要的角色.金属离子对生物体具有举足轻重的作用.一方面,金属离子参与构成生物体的许多重要分子,是机体不可缺少的组成部分.例如,光合作用所需的叶绿素中含有镁离子,动物呼吸作用所依赖的血红素中含有亚铁离子,在蛋白质晶体数据库(PDB)中近1/3的蛋白质含有金属离子.另一方面,金属离子可作为调节因子,参与调节生物分子的性质和功能.例如,Na+,K+,Mg2+和Ca2+可以帮助机体维持血液的渗透压[1].一些金属离子还参与调节酶的活性.
分子形貌理论[2]不仅可以描述分子的大小和形状,还可以描述分子界面上电子密度的分布情况,具有广泛的使用价值.例如,通过分子形貌理论定义的分子体积和表面积与物质的蒸发热有较好的相关性,可用来分析溶剂效应[3];利用分子形貌,可生动形象地展示分子在相互作用过程中,形状和电子密度的动态变化情况[4-5];还可以通过分子形貌对一些化学反应做出具体的分析和研究[6-7].
水溶性的金属离子,对理解含金属离子生物分子的结构和功能有指导作用,而成为实验和理论研究的热点[3,8-12].本文以M(H2O)1~4(M = Li+,Na+,Be2+和Mg2+)为研究对象,讨论随着水分子数目的增加,体系几何结构及属于化学键上各物理量的变化.计算了各体系的连续结合能,分析了连续结合能与键心处PAEM绝对值(Dpb)之间的关系.
1 模型与理论方法
分子中单电子受到的作用势(Potential Acting on an Electron in a Molecule,PAEM)[13-14]是衡量分子中单电子受到分子中其余电子和原子核的相互作用势能大小的物理量,可用式(1)表示:
.
(1)
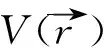

图1 Be2+H2O分子平面内单电子作用势的三维立体图Fig.1 The 3-dimensional graph of PAEM when electron runs on the molecular plane of Be2+H2O
分子的PAEM作用势为标量函数,且数值都为负.通常情况下,当电子逐渐靠近原子核时,PAEM作用势逐渐降低,反之,PAEM逐渐升高,且逐渐接近于零.在分子周围空间存在一些特征点,在这些点处,电子的PAEM作用势等于该分子第一电离能的负值.我们称这些点为电子运动的经典转折点.所有这些特征点的集合构成一特征界面,将空间划分成分子内和分子外两部分,该界面称为分子的内禀特征轮廓.将分子的电子密度附着在分子的内禀特征轮廓上,即可获得分子的分子形貌(molecular face,MF).
2 结果与讨论
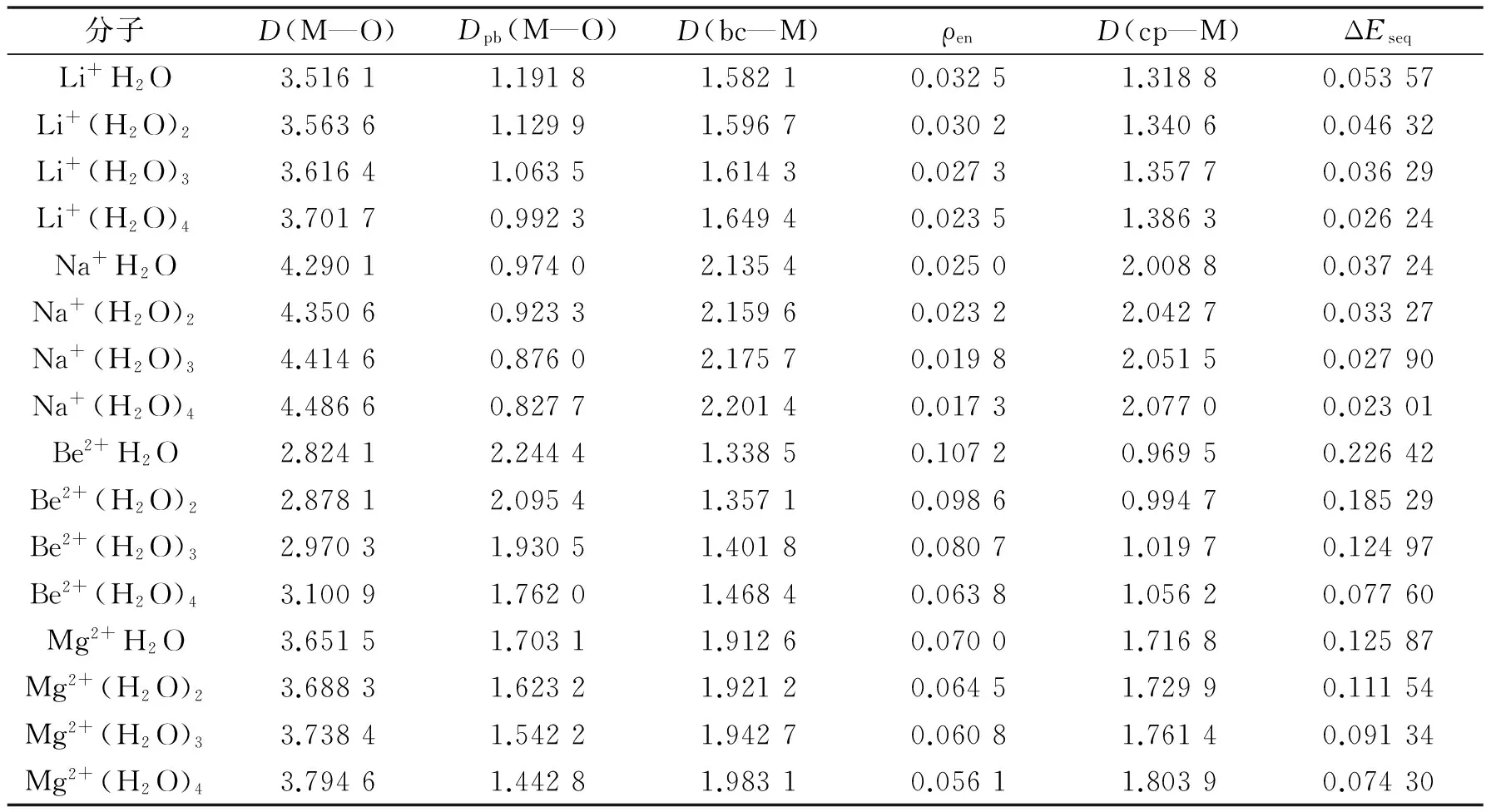
利用Gaussian 09软件包在MP2/6-311++G(3df,3pd)水平下,对所有体系进行了几何结构优化.计算结果表明:含相同水分子数的不同水合金属离子具有类似的几何结构,主要区别是金属离子与水分子中氧原子的距离不同,示于图2.从图2可以看出,各个团簇分子具有较好的对称性,随水分子数目的增加对称性依次为C2v,D2d和D3.
图2还展示了各个体系的分子形貌图,图中的颜色随电子密度的增大而变深.从图2可看出,在内禀特征轮廓上,电子密度的最大值出现氧原子附近,而最小值出现在金属离子附近.这表明,体系的电子富集于氧原子周围,而在金属离子周围出现的概率比较小.当体系增加一个水分子时,水分子中的氧原子进攻体系分子形貌上电子密度最小值附近.例如,从图2可看出,当MH2O结合一个水分子变成M(H2O)2时,水分子中氧原子结合在MH2O分子的分子形貌电子密度最小值附近,即与金属离子结合.
为了分析随水分子数目的增加,水合金属离子性质的变化情况,我们使用CISD/6-311++G(d,p)方法,计算了金属离子与水分子中氧原子之间化学键(M—O)上的几个重要的物理量,包括M—O 的键长D(M—O);键心(bond center,bc)到金属离子的距离,D(bc—M);键心处的PAEM的绝对值,Dpb(M—O);化学键上电子密度临界点(critical point,cp)到金属离子的距离,D(cp—M);临界点处的电子密度ρen,列于表1中.另外,在CCSD(FULL)/6-311++G(3df,3pd)方法基组下,计算了每结合一个水分子时体系的连续结合能,ΔEseq.本文对计算的连续结合能做了BSSE校正.连续结合能可用式(2)表示:
ΔEseq=E[M(H2O)n]+E(H2O)-E[M(H2O)n+1]
(2)

图2 水合金属离子M(H2O)1~4的结构及分子形貌图Fig.2 The geometry structures and MFs of hydrated metal ions M(H2O)1~4
将价电子数相同的金属形成的化合物归为一类,即含Li+和Na+的化合物作为一类,而含Be2+和Mg2+的化合物作为一类.从表1中可知,Na+H2O中Na+—O的键长为4.290 1 a.u.大于Li+H2O中Li+—O的键长3.516 1 a.u..也就是说,当水分子数目相同时,Na+—O键长大于Li+—O键长.这是由于在相同配位数的情况下,Na+的离子半径大于Li+的离子半径.
当水分子数目相同时,Li+(H2O)n的Dpb(Li+—O)大于Na+(H2O)n的Dpb(Na+—O).例如,Li+(H2O)4的Dpb(Li+—O)为0.992 3 a.u.大于Na+(H2O)4的Dpb(Na+—O) 0.827 7 a.u..这是因为Li相比Na具有较大的电负性,对电子的吸引能力相对较强.以上所得规律,同样适用于比较分析Be2+及Mg2+形成的金属离子化合物.
键心和化学键上电子密度临界点所对应的点,分别为化学键上2个特殊的点,都可以将化学键划分为两部分.通过表1可知,对所有体系,D(bc—M)都大于D(cp—M).以Li+H2O为例,D(bc—M)为1.582 1 a.u.,而D(cp—M)为1.318 8 a.u..即通过键心划分分子体系,金属离子可具有较大的空间.
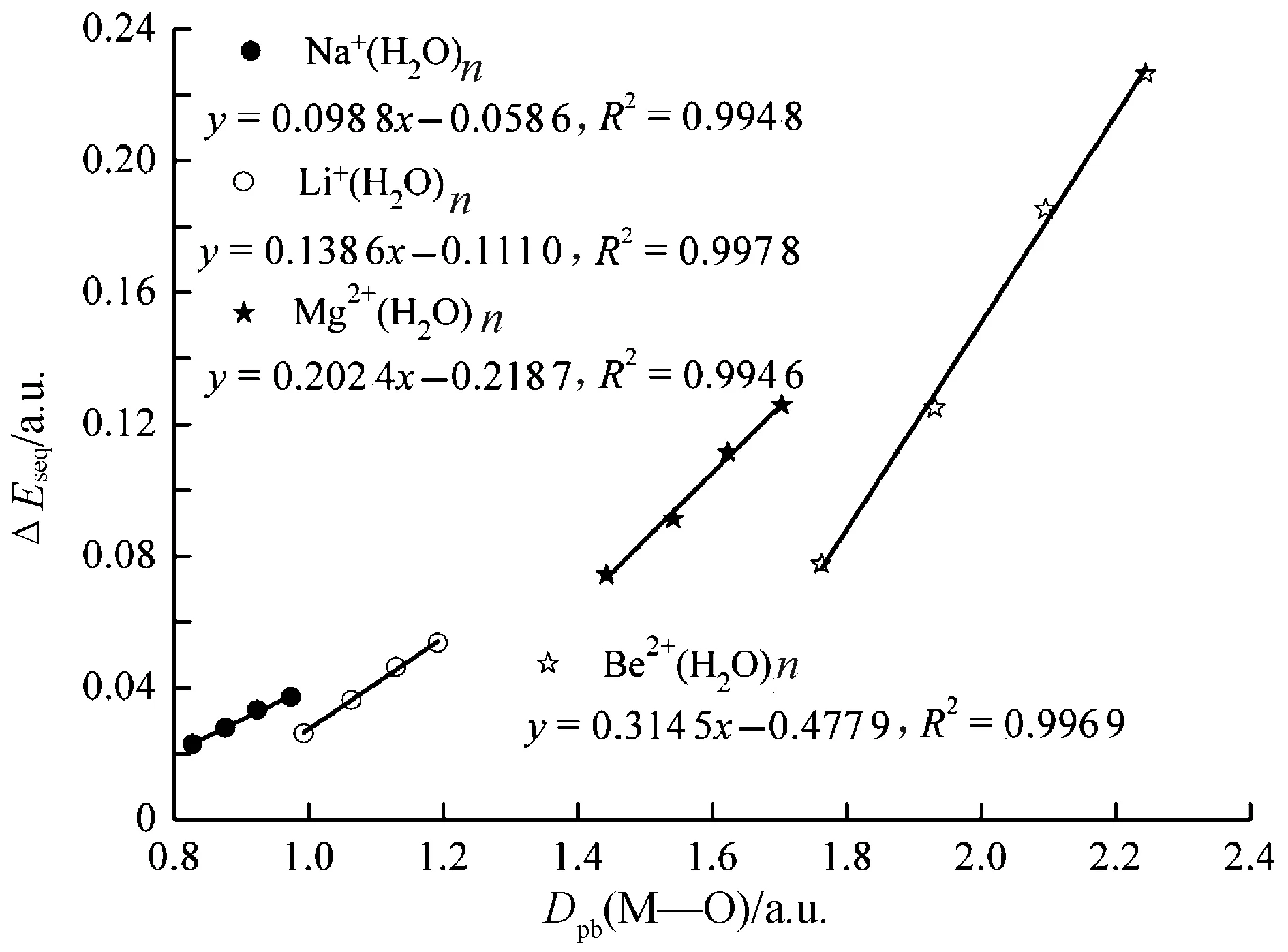
图3 金属离子与水分子中氧原子之间的Dpb与连续结合能之间的相关性Fig.3 Correlation of Dpb between metal ion and oxygen atom in water versus sequential binding energy
通过分析含锂离子的体系,发现:Li+(H2O)1~4随着水分子数目增加,M—O键长从3.516 1 a.u.增加到3.701 7 a.u.,Dpb(M—O)值从1.191 8 a.u.减小到0.992 3 a.u.,电子密度临界点处电子密度从0.032 5 a.u.减小到0.023 5 a.u.,连续结合能从0.053 57 a.u.减小到0.026 24 a.u..对含有相同金属离子的体系,随着水分子数目的增加,M—O键逐渐变长,Dpb(M—O)逐渐减小,键上电子密度临界点处的电子密度也逐渐减小,连续结合能逐渐降低.图3给出了金属离子体系中Dpb(M—O)与连续结合能之间的线性关系图.从图3可以看出,对含相同金属离子的体系,Dpb(M—O)与连续结合能之间的线性相关系数的平方R2都在0.99以上,两者有着很好的相关性.
综上所述,M—O键长,Dpb(M—O)的值,键上电子密度临界点处的电子密度值以及连续结合能,四者之间具有一定的线性关系.4个物理量都可用来分析各个体系中金属离子与水分子结合的强弱程度.
3 结 论
基于分子中单电子受到的作用势,绘制了水合金属离子的分子形貌.结果表明:在各个体系的内禀特征轮廓上,电子密度最大值出现在氧原子附近,而最小值出现在金属离子附近.并且指出,随水分子数目的增加,金属离子与水分子中氧原子之间化学键上,键心处PAEM值逐渐变大,PAEM势垒逐渐升高,电子密度临界点处电子密度值逐渐减小,连续结合能也逐渐减小.三者存在着一定的线性关系.为进一步讨论金属离子的性质,提供一定理论依据.
[1] PAGE M J,DI CERA E.Role of Na+and K+in enzyme function[J].Physiological Reviews,2006,86(4):1049-1092.
[2] YANG Z Z,ZHAO D X,WU Y.Polarization and bonding of the intrinsic characteristic contours of hydrogen and fluorine atoms of forming a hydrogen fluoride molecule based on an ab initio study[J].The Journal of Chemical Physics,2004,121(8):3452-3462.
[3] DAUB C D,ÅSTRAND P O,BRESME F.Lithium ion-water clusters in strong electric fields:A quantum chemical study[J].The Journal of Physical Chemistry A,2015,119(20):4983-4992.
[4] DING Y L,LI E B,GONG L D.Dynamic changing features of the molecular face for interaction of a rare gas atom with a hydrogen molecule[J].International Journal of Quantum Chemistry,2012,112(12):2515-2524.
[5] ZHAO D X,YANG Z Z.How the molecular face and the interaction vary as H atom approach H2molecule[J].Theoretical Chemistry Accounts,2014,133(11):1574-1-1574-12.
[6] YANG Z Z,DING Y L,ZHAO D X.Theoretical analysis of gas-phase front-side attack identity SN2(C) and SN2(Si) reactions with retention of configuration[J].The Journal of Physical Chemistry A,2009,113(18):5432-5445.
[7] ZHANG M B,YANG Z Z.Computational study on the reaction CH2CH2+F→CH2CHF+H[J].The Journal of Physical Chemistry A,2005,109(21):4816-4823.
[8] KREKELER C,HESS B,DELLE SITE L.Density functional study of ion hydration for the alkali metal ions (Li+,Na+,K+) and the halide ions (F-,Br-,Cl-)[J].The Journal of Chemical Physics,2006,125(5):054305-1-054305-7.
[9] RAO J S,DINADAYALANE T,LESZCZYNSKI J,et al.Comprehensive study on the solvation of mono-and divalent metal cations:Li+,Na+,K+,Be2+,Mg2+and Ca2+[J].The Journal of Physical Chemistry A,2008,112(50):12944-12953.
[10] 宫利东,任伟贺,杨忠志,等.应用从头计算和ABEEMσπ极化力场方法对MLn(M=Na+/K+;L=NMA,H2O;n=1~6)的研究[J].中国科学:化学,2016,46(1):114-125.
[11] LI X,YANG Z Z.Study of lithium cation in water clusters:Based on atom-bond electronegativity equalization method fused into molecular mechanics[J].The Journal of Physical Chemistry A,2005,109(18):4102-4111.
[12] 宫利东,刘燕,杨忠志.铝离子水分子团簇Al3+(H2O)n(n=1~6)性质的理论研究[J].辽宁师范大学学报(自然科学版),2010,33(02):209-212.
[13] ZHAO D X,YANG Z Z.Investigation of the distinction between van der Waals interaction and chemical bonding based on the PAEM-MO diagram[J].Journal of Computational Chemistry,2014,35(13):965-977.
[14] ZHAO D X,GONG L D,YANG Z Z.The relations of bond length and force constant with the potential acting on an electron in a molecule[J].The Journal of Physical Chemistry A,2005,109(44):10121-10128.
[15] ZHAO D X,YANG Z Z.Theoretical exploration of the potential and force acting on one electron within a molecule[J].The Journal of Physical Chemistry A,2014,118(39):9045-9057.
StudyonM(H2O)1~4(M=Li+,Na+,Be2+andMg2+)basedonthemolecularfacetheory
ZHAODongxia,ZHUZunwei,YANGZhongzhi
(School of Chemistry and Chemical Engineering, Liaoning Normal University, Dalian 116029, China)
The interaction mechanism between metal ions and water molecules is helpful for the understanding of the structures and functions of bio-molecules containing metal ions.We drew the molecular faces of M(H2O)1~4( M = Li+,Na+,Be2+and Mg2+) based on the Potential Acting on an Electron in a Molecule,PAEM.The electron density has the maximum around the oxygen atom while the minimum around the metal ions on the molecular intrinsic characteristic contour of all the model molecules.Then,we obtained the absolute of the PAEM at the bond center,Dpb,between the metal ion and oxygen atom.At the same time,we obtained electron density at the critical point between them.We also calculated sequential binding energies which can be used to characterize the strength of interaction between the metal ion and the water molecule.It can be found that theDpbs are in good correlation with their respective binding energies.This paper may be valuable for study on interactions between metal ions and other molecules.
PAEM;molecular face;Dpb;sequential binding energy;hydrated metal ions
O641.121
:A
2017-05-18
国家自然科学基金资助项目(21473083;21133005)
赵东霞(1971- ),女,河南长垣人,辽宁师范大学教授,博士,博士生导师.
1000-1735(2017)03-0337-05
10.11679/lsxblk2017030337
猜你喜欢
发明与创新·中学生(2023年2期)2023-01-09 03:50:05
椰城(2021年12期)2021-12-10 06:08:52
国防科技大学学报(2020年6期)2020-12-07 09:25:48
测绘通报(2019年11期)2019-12-03 01:47:34
赤峰学院学报·自然科学版(2019年5期)2019-09-10 07:22:44
测绘学报(2018年1期)2018-02-27 02:23:07
物理学报(2017年21期)2017-11-10 08:25:38
腐蚀与防护(2016年7期)2016-09-14 09:30:56
考试周刊(2016年60期)2016-08-23 07:42:47
考试周刊(2016年48期)2016-06-29 18:16:35
