
婴儿型Sandhoff病1例报告并文献复习
2017-09-16 06:30:07詹丽萍李栋方李平甘梁立阳罗向阳
临床儿科杂志 2017年9期
詹丽萍 李栋方 李平甘 梁立阳 罗向阳 黄 科
中山大学孙逸仙纪念医院儿科(广东广州 510120)
婴儿型Sandhoff病1例报告并文献复习
詹丽萍 李栋方 李平甘 梁立阳 罗向阳 黄 科
中山大学孙逸仙纪念医院儿科(广东广州 510120)
目的探讨婴儿型Sandhoff病的临床表现、诊断及治疗。方法回顾分析1例婴儿型Sandhoff病患儿的临床资料,并复习相关文献。结果1岁2个月女性患儿,有神经运动发育倒退、顽固性抽搐;父母为近亲婚配;眼底镜检查见眼底红斑;头颅磁共振成像示左侧脑桥可见一小点状长T2等T1异常信号影,脑白质水肿、弥漫性脱髓鞘改变;染色体核型未见异常;染色体微阵列提示多条染色体发生大片段纯合子;二代基因测序提示HEXB基因外显子11存在c.1263_1268delTGAAGT:P.(Glu422_Val423del)缺失突变及内含子13存在c.1614_2A>G:P?剪切突变,父母各携其一。白细胞HexA、HexA & HexB酶活性分别为84、112 nmol/(mg·h);确诊为婴儿型Sandhoff病。治疗采用丙戊酸钠、左乙拉西坦联合抗癫痫及糖皮质激素,患儿抽搐发作次数逐渐减少,反应较前好转;随访5个月,病情平稳无进展,无抽搐发作。患儿母亲再次妊娠,并于孕21+6周时行羊水穿刺检查,结果提示胎儿与患儿存在相同的突变。结论Sandhoff病是一种罕见的遗传性溶酶体病,主要表现为进行性神经功能损害,目前缺乏有效的治疗方法,基因检测有助确诊及产前诊断。
神经节苷脂累积病; Sandhoff病; HEXB基因; 婴儿型
GM2神经节苷脂沉积症(monosialoganglioside 2 gangliosidoses)是一种常染色体隐性遗传性溶酶体病,Sandhoff病为GM2神经节苷脂沉积病其中一种类型。由于HEXB基因突变导致β-氨基己糖苷酶同工酶A(β-hexosaminidase A,HexA)和B(HexB)共同缺陷,最终导致Sandhoff病。现就1例婴儿型Sandhoff病患儿的临床特点、诊断及治疗方法结合文献进行分析探讨。
1 临床资料
患儿,女,1岁2个月,因“反复发作性抽搐1个月,发作次数增多伴发热3天”入中山大学孙逸仙纪念医院儿科神经专科就诊。患儿G2P2,足月顺产,出生体质量3.8 kg,出生后无窒息及病理性黄疸史。患儿自幼对声响敏感,易出现惊跳;3个月抬头;4个月“yi ya”发音;8个月坐;9月可扶站、发“ma ma”双音节。9个月开始出现发育倒退,四肢活动减少,独坐时间缩短。13个月始不能抬头,不能坐,不会说话,哭声细弱,对周围亲人无反应,伴反复抽搐,表现为流涎,嘴角向右抽动,频繁眨眼,右侧手指抖动,无发绀,无牙关紧闭,呼之不应,持续数分钟至数小时不等,发热时抽搐频率增加。14个月时出现吞咽困难,易呛咳。患儿体质差,痰多,曾多次因呼吸道感染住院。父母近亲婚配(三代旁系),患儿父母及一胞姐均体健。家族中无类似病史。入院体格检查:头围37 cm,体质量10 kg,身长86 cm,神志清楚,表情呆滞,反应差,不能追光追物,对声音刺激有惊跳反应。皮肤白,头发黄、细,特殊面容,前囟未闭,稍隆起,约2.5 cm×3.0 cm。舌体肥厚。肺部可及大量哮鸣音,心脏听诊未见异常。腹部平软,肝肋下1 cm,质软,边缘锐利。脾肋下未及。四肢肌张力增高,双侧膝反射、跟腱反射亢进。踝阵挛阳性,双侧巴氏征(+),脑膜刺激征均阴性。入院实验室检查:血、尿、粪常规、肝肾功能、心肌酶、乳酸、氨基酸和酰基肉碱谱分析、尿液有机酸分析、寄生虫七项均未见异常。脑脊液压力高,260 cmH2O,脑脊液常规、免疫性脑炎检测未见异常,脑脊液生化示乳酸脱氢酶(LDH)88 U/L,余未见异常。眼底镜检查示双眼底黄斑区可见樱桃红斑,见图1。听性脑干诱发电位:听觉中枢传导时间延长。脑电图示双侧导联散在中等波幅尖波,右侧额叶明显。彩色多普勒超声心动图:二尖瓣脱垂并轻度反流,三尖瓣轻微反流。腹部B超、泌尿系B超、脊柱正侧位片未见异常。头颅磁共振成像(MRI)示左侧脑桥可见一小点状长T2等T1异常信号影,脑白质水肿、弥漫性脱髓鞘改变。见图2。
经患儿家属同意,收集患儿、胞姐、父母外周血各4 mL,送广东省广州市妇女儿童医疗中心行外周血白细胞Hex A、Hex A&B酶活性检测,方法参照文献[3]。酶学分析结果提示,患儿白细胞内HexA酶活性为84 nmol/(mg·h)、Hex A&B酶活性为112 nmol/(mg·h),均显著低于正常值。胞姐的HexA酶活性稍低于正常值,Hex A&B酶活性正常;父母两种酶的活性均正常,见表1。

表1 患儿及直系亲属血白细胞内Hex酶活性(nmol·mg-1·h-1)

图1 患儿黄斑区眼底红斑
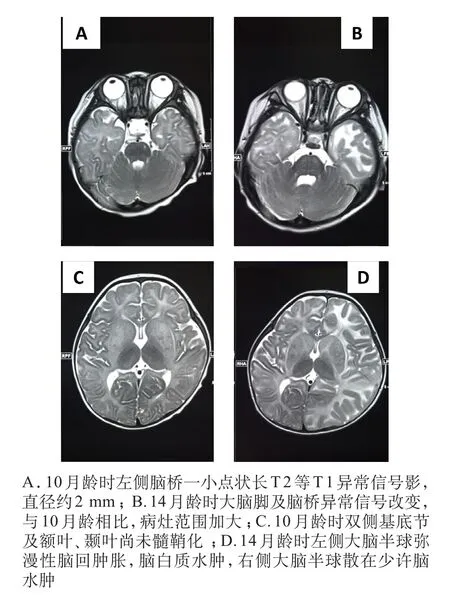
图2 患儿头颅MRI
患儿染色体核型分析未见异常。进一步采用Affymetrix公司配套检测试剂盒,使用CytoScanHD/CytoScan 750 K进行全基因组范围扫描。结果显示,患儿多条染色体发生大片段纯合子(>3 Mb),片段长度总和约为127.5 Mb,占常染色体基因组片段总长度的4.5%。
另抽取患儿及家系成员外周静脉血各2 mL,乙二胺四乙酸二钠(EDTA)抗凝;送广州市金域医学检验中心,通过高通量测序技术,对基因全外显子区进行直接测序,与参考序列(hg19)进行比较。结果发现,患儿HEXB基因外显子11存在c.1263_1268delTGAAGT:p.(Glu422_Val423del)缺失突变及内含子13存在c.1614-2A>G:p?剪切突变。其父亲携带c.1263_1268delTGAAGT:p. (Glu 422_Val423del),其母亲携带c.1614-2A>G:p?,进一步验证胞姐,发现其存在与父亲相同的突变位点。
患儿入院后予丙戊酸钠及左乙拉西坦联合抗癫痫治疗,积极抗感染、降颅压对症支持治疗,同时予地塞米松0.3 mg/(kg·d)静脉注射治疗及神经康复理疗,患儿抽搐逐渐减少,反应较前好转,哭声较前响亮,左侧肢体活动增多,偶有抬头动作,呛咳次数较前减少,前囟张力逐渐降低。确诊为Sandhoff病后,因缺乏有效治疗方法,患儿住院1个月后病情稳定,家属要求出院,改为口服泼尼松1 mg/(kg·d),门诊随诊。患儿使用激素1个月后,病情平稳,无抽搐发作,反应可,会笑,四肢活动增多,仍有惊跳及呛咳,不会说话,不能坐。现患儿19月龄,激素已逐渐减停,无抽搐发作,病情平稳,继续观察随访中。
在患儿确诊为Sandhoff病时,其母亲已怀有身孕,胎儿性别为女性,约20周,建议其行胎儿产前诊断。遂于21+6周时至外院行羊水穿刺,根据本例患儿HEXB基因突变点及邻近核酸序列对胎儿羊水进行检测,结果提示胎儿与患儿存在相同的突变,予引产处理。先证者家系图见图3。

图3 患儿家系图
2 讨论
Sandhoff病为常染色体隐性遗传病,由Konrad Sandhoff于1968年首先报道[4],根据发病年龄分为3种类型:婴儿型、青少年型、成人型。其发病率低,以散发为主,男女无差异,国外报道加拿大萨喀温省发病率较高[5]。Nicholas等[6]总结美国73例GM2神经节苷脂沉积症患者,发现10例青少年型Sandhoff病患者存在巴基斯坦血统。近亲结婚是该病发病的重要因素。本例患儿的父母为三代旁系近亲结婚,胞姐携带其父亲的突变基因,胞妹携带跟患儿同样的突变基因。国外亦有文献报道83%的患者有近亲结婚的家族史[7]。因缺乏对该病的认识及确诊方法,常被误诊为脑型瘫痪[8,9]。本例患儿通过染色体微阵列及基因分析,为诊断提供了线索,进一步通过己糖胺酶活性的检测明确了疾病的诊断。
婴儿型Sandhoff病的临床表现为进行性神经功能损害、听觉过敏、肌张力减低、双眼底视网膜黄斑区樱桃红斑、癫痫。症状多于3~9个月开始出现,如不能抬头,不能坐,肢体运动减少,肌张力减低,全身瘫软,语言丧失,失明,吞咽困难等。本例患儿自幼易出现惊跳动作,9个月开始出现运动发育倒退,后伴频繁抽搐发作,四肢肌张力增高。该患儿彩色多普勒超声心动图检查提示存在二尖瓣脱垂伴轻度反流,三尖瓣轻度反流,国外亦有文献报道患者存在心脏瓣膜病[10],其他异常均有见报道,如支气管及肺发育异常、并指畸形及少见的摇椅足[11-13]。该病进展较快,晚期症状重,一般于3~5岁以内死亡,死因以原发病为主,其他包括肺炎、癫痫[2]。
婴儿型Sandhoff病神经系统影像学检查并无特异性的改变,有报道CT检查提示两侧丘脑均匀高密度影,而MRI见T2WI低信号可能是该病的一个重要特征,推测这与细胞内GM2神经节苷脂沉积导致钙异位沉积相关,亦可有脑皮质萎缩、胼胝体变薄、苍白球、壳核、小脑、脑干、尾状核异常信号[13]。本例患儿头颅影像学表现与以往报道的婴儿型Sandhoff病的影像学特征存在显著差异。头颅MRI提示有明显的脑水肿表现,而脑脊液压力增高明显,但常规及生化等检查未见异常,患儿出现急性脑病症状,考虑可能与出现癫痫持续状态频繁抽搐有关,也不排除急性非细菌性感染导致中毒性脑病可能。
HXEB基因位于5q13上,由14个外显子组成,有文献报道迄今大概存在50种与Sandhoff病相关的不同基因突变类型,包括错易突变、无义突变、小片段缺失、小片段插入、小片段插入缺失、大片段缺失、剪切突变[1]。本例患儿存在第11外显子c.1263_1268delTGAAGT微小缺失突变及第13内含子c.1614-2A>G剪切突变,有报道在1名中国患者中检测到前者的纯合突变,体外蛋白活性检测结果显示,该突变使总Hex蛋白活性降低至正常对照样本的15%;后者未见文献报道,千人基因组计划数据库中均未见检出[14]。c.1263_1268delTGAAGT突变导致所编码的蛋白质第422位谷氨酸和第423位缬氨酸缺失,c.1614-2A>G剪切突变导致蛋白的变化,目前尚不清楚。结合其下游产物水平及患儿临床表现,可以推断患儿的两个突变为致病突变,导致疾病发生。其中第13内含子c.1614-2A>G剪切突变导致该疾病。
β-氨基己糖苷酶可分为β-氨基己糖苷酶A(HexA)和β-氨基己糖苷酶B(HexB)两种同工酶,HexA是由HEXA基因编码的α链和HEXB基因编码的β链组成的异二聚体,而HexB为两条相同的β链组成的同二聚体。HEXB基因突变导致β链缺陷,从而使HexA及HexB酶活性均降低[2],临床表现的严重程度常被认为与酶活性的高低相关。该先证者HexA、HexA&HexB两种酶活性均显著下降,患儿父母作为HEXB突变基因的携带者,两种酶的活性均为正常值的下限,胞姐HexA酶活性低于正常值,而HexA&HexB酶活性则为正常值下限,考虑可能在基因表达的过程中,携带者突变的基因下调了两种蛋白的表达,从而导致酶活性在正常值下限,甚至低于正常值;同时鉴于患儿父母为近亲结婚,亦可能存在某些基因影响了胞姐基因的表达。
迄今Sandhoff病无特效的治疗方法,主要是支持对症治疗。有报道采用美格鲁特(Miglustat)联合生酮饮食治疗1例青少年型Sandhoff病患儿有一定的治疗效果[15],本例患儿采用糖皮质激素合并抗癫痫药物治疗后,病情有所好转,抽搐控制,随访5个月疾病无进展。
遗传咨询及产前诊断在预防该病的发生尤为重要,在本例患儿及其父母基因型明确的情况下,其母亲在孕21+6周时行羊水穿刺,通过羊水细胞HEXB基因分析证实胎儿存在与患儿相同的突变基因,予引产处理。
[1]Zhang W, Zeng H, Huang Y, et al. Clinical, biochemical and molecular analysis of five Chinese patients with Sandhoff disease [J]. Metab Brain Dis, 2016, 31(4):861-867.
[2]Bley AE, Giannikopoulos OA, Hayden D, et al. Natural history of infantile G (M2) gangliosidosis [J]. Pediatrics,2011, 128(5): e1233-e1241.
[3]黄永兰, 谢婷, 郑纪鹏, 等. 青少年型Sandhoff病一例临床特点及基因分析 [J]. 中华儿科杂志, 2014, 52(4):313-316.
[4]Giles L, Cooper A, Fowler B, et al. First trimester prenatal diagnosis of Sandhoff's disease [J]. Prenat Diagn, 1988, 8(3):199-205.
[5]Fitterer BB, Antonishyn NA, Hall PL, et al. A polymerase chain reaction-based genotyping assay for detecting a novel Sandhoff disease-causing mutation [J]. Genet Test Mol Biomarkers, 2012, 16(5): 401-405.
[6]Smith NJ, Winstone AM, Stellitano L, et al. GM2 gangliosidosis in a UK study of children with progressive neurodegeneration: 73 cases reviewed [J]. Dev Med Child Neurol, 2012, 54(2): 176-182.
[7]Karimzadeh P, Jafari N, Nejad BH, et al. GM2-Gangliosidosis(Sandhoff and Tay Sachs disease): Diagnosis and Neuroimaging Findings (An Iranian Pediatric Case Series) [J].Iran J Child Neurol, 2014, 8(3): 55-60.
[8]马秀伟, 蒲利华, 张月华, 等. GM2神经节苷脂沉积症的临床特征及诊断 [J]. 实用儿科临床杂志, 2008, 23(7):539-541.
[9]吴桐菲, 李溪远, 王峤, 等. 一例婴儿型Sandhoff病家系的基因诊断与产前诊断 [J]. 浙江大学学报(医学版),2013, 42(4): 403-410.
[10]Venugopalan P, Joshi SN. Cardiac involvement in infantile Sandhoff disease [J]. J Paediatr Child Health, 2002, 38(1):98-100.
[11]Nair PM, Bataclan F, Ganesh A. Sandhoff disease in an extreme preterm baby with bilateral syndactyly [J].Neurosciences (Riyadh), 2003, 8(4): 246-247.
[12]Abdul-Wahab A, Bessisso MS, Elsaid MF. Sandhoff disease (GM2 gangliosidoses) in a premature patient with bronchopulmonary dysplasia [J]. Neurosciences (Riyadh),2002, 7(3): 194-197.
[13]Beker-Acay M, Elmas M, Koken R, et al. Infantile type Sandhoff disease with striking brain MRI findings and a novel mutation [J]. Pol J Radiol, 2016, 81: 86-89.
[14]Zampieri S, Cattarossi S, Oller RA, et al. Sequence and copy number analyses of HEXB gene in patients affected by Sandhoff disease: functional characterization of 9 novel sequence variants [J]. PLoS One, 2012, 7(7): e41516.
[15]Villamizar-Schiller IT, Pabon LA, Hufnagel SB, et al.Neurological and cardiac responses after treatment with miglustat and a ketogenic diet in a patient with Sandhoff disease [J]. Eur J Med Genet, 2015, 58(3): 180-183.
Infantile Sandhoff disease: a case report and literature review
ZHAN Liping, LI Dongfang, LI Pinggan, LIANG Liyang,LUO Xiangyang, HUANG Ke (SUN Yat-sen Memorial Hospital, SUN Yat-sen University, Guangzhou, 510120, Guangdong,China)
ObjectiveTo summarize the clinical manifestations, diagnosis, and treatment of infantile Sandhoff disease.MethodsThe clinical data of one case with infantile Sandhoff disease were reviewed retrospectively. The related literatures were reviewed.ResultsThe girl aged 1 year and 2 months suffered from psychomotor regression and intractable convulsions.The parents were consanguineous marriage. The fundus microscopy showed fundus erythema. Brain magnetic resonance imaging showed an abnormal signal of long T2WI and identical T1WI at left pons, white matter edema, and diffuse demyelination. No abnormal karyotype was observed. A chromosome microarray suggested multiple large homozygous chromosomes segments. The second generation gene sequencing showed deletion of c.1263_1268delTGAAGT:P. (Glu422_Val423del) deletion in exon 11 and a shear mutation of c.1614_2A>G:P? in intron 13 of HEXB gene which were carried by her parents respectively . The activity of HexA, HexA & HexB were 84 and 112 nmol·mg−1·h−1, respectively. Finally, this girl was diagnosed of infantile Sandhoff's disease. After treatment with valproate, levetiracetam combined with antiepileptic and glucocorticoids, episodes of convulsions were decreased gradually, and the reaction was better than before. In 5 months of follow up, the condition was stable, and no progression and no seizures exist. Her mother got pregnant again and
an amniocentesis on her 21+6weeks of pregnancy,and results suggest that the fetus had the same mutation as this girl.ConclusionsSandhoff's disease is a type of rare hereditary lysosomal disease, characterized by progressive neurological impairment. Currently there are no effective treatments. Genetic testing is helpful in the diagnosis and prenatal diagnosis.
ganglioside disease; sandhoff disease; HEXB gene; infantile
2017-01-22)
(本文编辑:邹 强)
10.3969/j.issn.1000-3606.2017.09.015
广东省自然基金博士启动项目(No.2015A030310047)
李栋方 电子信箱:ldf201310@163.com
猜你喜欢
英语世界(2023年10期)2023-11-17 09:18:44
心电与循环(2021年4期)2021-11-29 02:41:56
中成药(2021年5期)2021-07-21 08:38:32
心电与循环(2020年3期)2020-06-18 13:43:12
上海包装(2019年2期)2019-05-20 09:10:52
中成药(2018年2期)2018-05-09 07:19:49
中国眼镜科技杂志(2016年17期)2016-10-24 08:36:30
数位时尚(幼儿教育)(2016年9期)2016-10-15 08:57:07
微生物与感染(2015年1期)2015-02-28 17:42:37
英语学习(2015年12期)2015-02-01 14:08:30
