
ANK1基因突变致遗传性球型红细胞增多症1例报告
2017-09-16 06:30:07万伍卿
临床儿科杂志 2017年9期
张 丹 万伍卿
湘雅二医院儿童医学中心(湖南长沙 410000)
ANK1基因突变致遗传性球型红细胞增多症1例报告
张 丹 万伍卿
湘雅二医院儿童医学中心(湖南长沙 410000)
目的探讨遗传性球型红细胞增多症(HS)的遗传学特征和诊治。方法回顾分析1例确诊HS患儿的临床资料,复习相关文献资料。结果5岁女性患儿,生后6个月始有溶血性贫血;孵育脆性试验阳性,血涂片及红细胞电镜见球型红细胞;DNA测序示杂合性stopgain SNV改变,确诊HS,拟于6岁后行脾切除术。结论HS为常染色体显性遗传为主的遗传病,主要表现为贫血、溶血、脾大等,早期诊治有赖于基因检测。
遗传性球型红细胞增多症; ANK1基因; 锚蛋白; 血影蛋白
遗传性球型红细胞增多症(hereditary spherocytosis,HS)是一种最常见的红细胞膜缺陷引起的慢性溶血性贫血,多见于以下基因的改变:ANK1、EPB3、ELB42、SPTA1和SPTB。常见的临床表现有贫血、溶血和脾肿大。由于HS本身存在临床严重程度、蛋白缺陷和遗传方式异质性,散发存在,易被漏诊和误诊。现报告1例新的ANK1基因突变,即杂合性stopgain SNV改变患儿的临床资料,并进行相关文献复习。
1 临床资料
女性患儿,5岁,因“发现贫血4年余,发热伴咳嗽2天”入院。患儿于出生6个月起有轻中度贫血,血红蛋白波动在80~90g/L。入院体检:体温36.5℃,脉搏93次/min,呼吸20次/min,神志清,精神可,全身皮肤轻度黄染,全身浅表淋巴结未触及肿大;双肺呼吸音清,未闻及干湿啰音;心律齐,心音有力;腹部平软,肝肋下2 cm,脾肋下4 cm。实验室检查:血常规白细胞6.45×109/L,中性粒细胞0.76,血红蛋白65g/L,红细胞2.22×1012/L,血小板215×109/L。尿常规:尿胆原35 μmol/L,尿胆红素-;总胆红素51.4 μmol/L,直接胆红素8.4 μmol/L;CD55、CD59无异常;Coomb’s试验阴性;红细胞渗透脆性孵育试验:孵育前0.5%(参考值0.4%~0.45%),孵育后0.7%(0.46%~0.59%)。腹部彩超:肝稍大(肋下约17mm),脾大(厚约41mm,肋下40 mm)。骨髓细胞学检查:骨髓增生明显活跃,红系占55.5%,其中晚幼阶段可见分裂相和双核现象,成熟红细胞大小不均,可见球型红细胞。血涂片:可见35%球型红细胞;红细胞电镜:部分红细胞呈球形,部分红细胞表面有微小绒毛状突起(图1)。
经医院医学伦理审核并获得知情同意后,抽取患儿及父母的静脉血各5 mL,由武汉康盛达医学检验中心进行基因检测。采用二代测序方法,用DNA提取试剂盒(QIAamp DNA Blood MiniKit,Qiagen)提取患儿基因组DNA,用Nandrop 2000分光光度计进行DNA定量检测。采用超声打断仪(Covariss2,Massachusetts,USA)将患儿基因组DNA随机打断成主带200~300 bp的片段。按照illumina标准建库方法构建DNA测序文库。采用IlluminaHiSeq X10高通量测序仪测序,用Illumina Pipeline software(version 1.3.4)进行数据读取,读取后利用SOAPsnp软件和Samtools pileup软件进行分析。同时应用SIFT在线预测工具和PolyPhen-2软件预测突变对蛋白质功能的影响。

图1 红细胞电镜表现
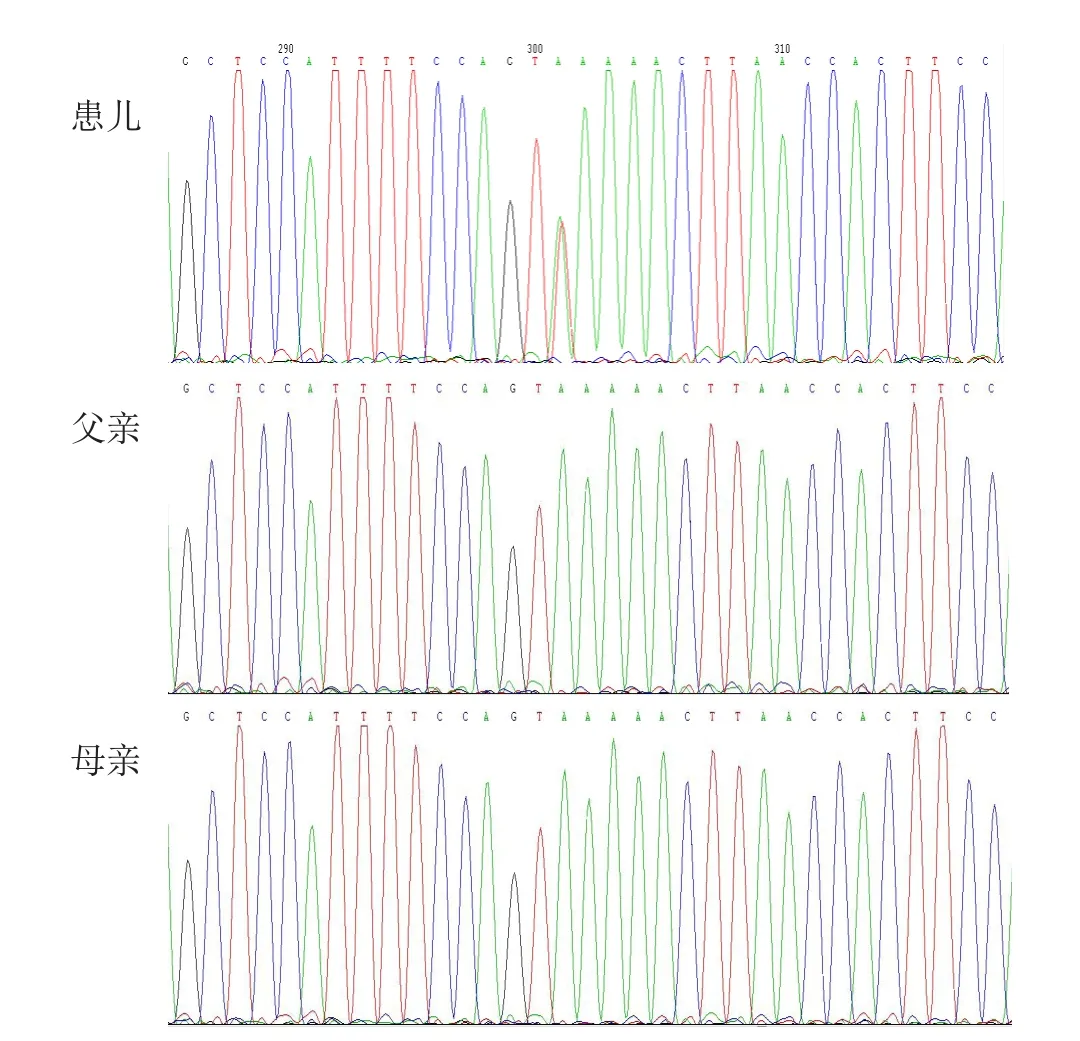
图2 基因测序图
基因检测结果:患儿ANK1基因呈A>AT杂合性改变,另一条链即T>TA改变;患儿父亲、母亲为正常野生型碱基A(图2)。该突变称为杂合性stopgain SNV改变,NM-000037:c.T389A:p.L130X。其具体氨基酸突变异常情况为:NM-00037:exon5:c.T389A:p.L130X,ANK1:NM-001142446:exon5:c.T488A:p.L163X,ANK1:NM-020475:exon5:c.T389A:p.L1130X,ANK1:NM-020476:exon5:c.T389A:p.L1。
患儿确诊为HS,予抗感染、补充叶酸等治疗,拟6岁后行脾切除治疗。
2 讨论
HS是最常见的遗传性红细胞膜紊乱,多见于北欧和北美,发病率为 1:5 000。国内以散发为主,据统计1978-2013年共2 034例HS患者,发病率男性为1.27/100 000,女性1.49/ 100 000,多见于中国东部和南部,以山东、北京、辽宁、河北、上海多见[1]。HS患者中约70%为显性遗传,25%为隐性遗传,5%为新生突变[2]。
HS为一种遗传性分子缺陷病,其发病机制至今仍未清楚。HS的形成与红细胞无法维持正常的双凹形,造成过早破坏有关,目前已知的抗红细胞弹性变形的结构是由蛋白质网络(骨架)、脂质双层(板层)和跨膜蛋白质相互交联形成[3]。目前已知的相关基因及其相应的编码蛋白有:①SPTA1基因(血影蛋白A链);②SPTB基因(血影蛋白B链);③ANK1基因(锚蛋白);④EPB3基因(蛋白AE1,或带3);⑤ELB42基因(蛋白4.2)。通过基因突变的频率递减顺序可排列为:ANK1,EPB3,SPTB,ELB42和SPTA1基因。HS的大部分病例与ANK1突变有关[4]。已有文献报道的HS中有60种ANK1突变,包括有7项错义突变。具有锚蛋白缺陷的球形红细胞增多症患者,临床严重程度取决于膜损失的程度,除了一个错义突变(HS04)无害以外,大多数ANK1突变是有害的,包括移码、剪接错误和无义突变[5]。本例患儿系新生突变,为生后ANK1基因损伤,表现为ANK1碱基位点突变,为获得性体细胞突变,主要见于球型细胞增多症1型,系常染色体显性遗传,可无明显家族史,一般有赖于基因检测诊断。
HS是一种具有临床严重程度、蛋白缺陷和遗传方式异质性的遗传性溶血性贫血。临床严重程度轻重不一,从无症状到危及生命的贫血[4]。这种变异性不仅与不同的分子缺陷相关,并与骨髓代偿有关。HS属于一种慢性溶血性贫血,主要特征为黄疸、脾大、红细胞球型改变、渗透脆性增加。新生儿期HS通常有症状,新生儿期发病时可有严重的贫血和黄疸,需要光疗、输血,甚至换血;婴儿期除了轻中度贫血外,常无其他症状;幼儿及年长儿发病时,主要症状为轻度黄疸和贫血,诱因如过度劳累、受凉感冒等抵抗力下降时可引起黄疸加重,伴随症状有发热、呕吐、腹痛、肝脾增大及压痛、乏力、心跳加速、气促等;成年期的HS一般临床表现较典型,贫血、黄疸和脾肿大多见。外周血涂片存在球形红细胞和红细胞渗透脆性增加是本病的特点。本例患儿生后即有贫血,感染后有溶血、脾大,血涂片、骨髓涂片、红细胞电镜均可见球形红细胞,红细胞渗透脆性增加支持HS诊断,无明显家族史,经过血液系统遗传疾病基因筛查确诊为HS,考虑系ANK1基因损伤所致新生突变。
约75%的HS患者有明确的家族病史,有典型的病史和体格检查时诊断大多不难,目前主要的实验室方法有:血常规、外周血涂片、红细胞渗透脆性试验、酸化甘油溶解试验、伊红-马来酰亚胺结合试验、十二烷基硫酸钠一聚丙烯酰胺凝胶电泳法以及基因诊断。各个实验室方法各有利弊,需要注意的是正常的红细胞渗透脆性试验的结果并不能排除诊断HS,可能有10%~20%的HS患者该试验可为阴性。主要鉴别诊断自身免疫性溶血、遗传性口形红细胞增多症以及相关的钠和钾离子通透性异常的疾病。HS患者必须与遗传性口形红细胞增多症区分,因为若为后者脾切除术后血栓形成风险增加,切脾后口形红细胞内皮黏附性增高,可出现血液高凝状态而致严重血栓形成,因此切脾需慎重。
HS常见的并发症有:再生障碍性危机、胆结石、血色素沉着症,罕见并发症有生长衰竭、严重情况下的骨髓扩展和骨骼畸形、腿部溃疡等。再生障碍性危机是一种较常见的并发症,可作为HS患者首发症状,主要与人细小病毒B19感染有关,表现为贫血、黄疸、脾大、发热、严重的苍白,通常在脸颊有斑丘疹(掌掴脸颊),并常有腹泻和呕吐,血涂片有球形红细胞,轻度血小板减少和白细胞减少伴感染,丙种球蛋白的使用有利于缩短病程[6]。胆结石是最常见的并发症。血色素沉着症见于HS患者在输血或脾切除后,多见杂合状态的患者[7]。
目前关于HS的治疗主要有注意休息,加强营养,多补充新鲜水果蔬菜,避免感染,适当补充叶酸。补充叶酸对于有慢性溶血的重度HS尤其重要。脾切除术是目前治疗HS最有效的方法,可防止溶血,避免胆结石,避免脾创伤破裂的危险。脾切除最常见和危险的并发症是凶险的脾切除后感染(OPSI)[8]。OPSI表现为有荚膜的细菌,如肺炎链球菌,广泛感染以后,引起多脏器功能衰竭,病死率高达50%[9]。关于脾切除术的手术时机指南推荐最好在6岁以后,青春期前进行。本例患儿目前已确诊为HS,予口服叶酸,嘱避免感染,定期复查血常规,监测贫血、溶血情况,避免再障危象、胆石症等并发症的发生,并计划于6岁后行腹腔镜下脾切除术。
HS是一种遗传性的分子缺陷病,随着对基因位点的深入研究和基因诊断的开展,漏诊率和误诊率降低。新的突变位点的发现加深了对HS致病基因的理解。有HS家族史的孕妇需作基因检测,做好产前诊断,以免影响下一代。
;
[1]Wang C, Cui Y, Li Y, et al. A systematic review of hereditary spherocytosis reported in Chinese biomedical journals from 1987 to 2013 and estimatiom of prevalence of the disease using a disease model [J]. Intractable Rare Dis Res, 2015,4(2): 76-81.
[2]卢新天.遗传性球形红细胞增多症发病机制、诊断及治疗进展[J].中国小儿血液与肿瘤杂志, 2009, 14(6): 243-245.
[3]Iolascon A, Miraglia del Giudice E, Perrotta S, et al.Hereditary spherocytosis: from clinical to molecular defects[J]. Haematologica, 1998, 83(3): 240-257.
[4]Bogusławska DM, Heger E, Listowski M,et al. A novel L1340P mutation in the ANK1 gene is associated with hereditary spherocytosis [J]. Br J Haematol, 2014, 167(2):269-272.
[5]Park J, Jeong DC, Yoo J,et al.Mutational characteristics of ANK1 and SPTB genes in hereditary spherocytosis [J]. Clin Genet, 2016, 90(1): 69-78.
[6]Alavi S Md, Arabi N Md, Yazdi MK Md, et al. Hereditary spherocytosis unmasked by human parvovirus B19 induced aplastic crisis in a family [J]. Iran J Med Sci, 2015, 40(5):461-464.
[7]陈宝鹤,杨小勇,李文美.腹腔镜与开腹脾切除治疗遗传性球型红细胞增多症的近期疗效观察[J].吉林医学,2015, 36(1): 50-51.
[8]Wagener FA, Eggert A, Boerma OC, et a1. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase [J]. Blood, 200l, 98(6): 1802-1811.
[9]金洁,童茵.脾切除术治疗贫血的再评价[J].内科理论与实践, 2010, 5(4): 310-313.
Hereditary polycythemia caused by mutation of ANK1 gene: a case report
ZHANG Dan, WAN Wuqing (Children’s Medical Center of the Second Xiangya Hospital, Changsha 410000, Hunan, China)
ObjectiveTo explore the genetic characteristics, diagnosis, and treatment of hereditary spherical polycythemia (HS).MethodsThe clinical data of one case of HS was analyzed retrospectively, and related literatures were reviewed.ResultsThe 5-year-old girl presented hemolytic anemia from 6 months old. Incubation of fragility tests was positive. Blood smears and red cell electron microscopy showed spherical red blood cells. DNA sequencing showed alterations in heterozygosity of stopgain SNV. The girl was diagnosed was HS, and was scheduled to undergo splenectomy at 6 years old.ConclusionsHS is an autosomal dominant genetic disease, mainly manifested as anemia, hemolytic anemia, and splenomegaly. The early diagnosis depends on genetic testing.
hereditary spherical erythrocytosis; ANK1 gene; ankyrin; spectrin
2017-01-21)
(本文编辑: 梁 华)
10.3969/j.issn.1000-3606.2017.09.014
万伍卿 电子信箱:wanwuqing65@163.com
猜你喜欢
中国现代医生(2022年19期)2022-11-04 10:13:29
粘接(2022年8期)2022-08-19 07:47:14
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
建材发展导向(2022年3期)2022-04-19 12:51:24
基层中医药(2021年8期)2021-11-02 06:24:54
制造技术与机床(2019年10期)2019-10-26 02:47:54
北京航空航天大学学报(2017年12期)2017-04-23 08:31:43
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:37:12
现代机械(2016年6期)2016-12-20 11:05:03
大型铸锻件(2015年1期)2016-01-12 06:33:06
