
罗格列酮对小鼠高胆固醇血症的治疗作用、肝脏胆汁酸代谢影响及其机制探讨
2017-08-30 10:06:04何芋岐凌蕾杜艺玫马菲菲朱春媛周若雨茆芮婕周旭美王玉和鲁艳柳
山东医药 2017年31期
何芋岐,凌蕾,杜艺玫,马菲菲,朱春媛,周若雨,茆芮婕,周旭美,王玉和,鲁艳柳
(1遵义医学院国家级药学实验教学示范中心药学实验室,贵州遵义 563099;2遵义医学院基础药理教育部重点实验室暨特色民族药教育部国际合作联合实验室;3遵义医学院附属医院)
罗格列酮对小鼠高胆固醇血症的治疗作用、肝脏胆汁酸代谢影响及其机制探讨
何芋岐1,2,凌蕾1,2,杜艺玫1,2,马菲菲1,2,朱春媛1,2,周若雨1,2,茆芮婕1,2,周旭美1,王玉和3,鲁艳柳2
(1遵义医学院国家级药学实验教学示范中心药学实验室,贵州遵义 563099;2遵义医学院基础药理教育部重点实验室暨特色民族药教育部国际合作联合实验室;3遵义医学院附属医院)
目的 观察罗格列酮对小鼠高胆固醇血症的治疗作用、肝脏胆汁酸代谢影响,并探讨其可能机制。方法 36只小鼠随机分为3组各12只,干预组和模型组均制备高胆固醇血症模型,并分别灌胃罗格列酮羧甲基纤维素钠混悬液、空白羧甲基纤维素钠溶液,持续给予高脂饲料;正常组每日灌胃等体积羧甲基纤维素钠溶液,始终给予普通饲料。观察各组组织病理学及血生化指标[血糖、血清总胆固醇(TC)、低密度脂蛋白胆固醇(LDL-C)、谷丙转氨酶(ALT)、谷草转氨酶(AST)]变化。液质联用仪分析肝脏组织中胆汁酸丰度。另检测肝脏或肠组织中Cyp7a1、Cyp8b1、FXR、SHP、Lrh1、Hnf4α、Fgf15、Fgfr4、JNK1/2、ERK1/2基因。结果 模型组肝细胞中充斥大量脂肪泡,干预组细胞内脂肪泡接近正常组。与模型组比较,干预组TC、LDL、血糖水平降低,ALT水平升高,P均<0.05。模型组TCA显著下降,但干预组没有能逆转其下降的趋势;模型组DCA无太大变化,但干预组DCA急剧上升,远远超过对照组。与模型组比较,干预组JNK1、Erk2、Cyp7a1、FXR表达升高,SHP表达降低,P均<0.05。结论 罗格列酮对小鼠高胆固醇血症有治疗作用,且影响肝脏胆汁代谢,其机制可能为调控胆汁酸代谢通路基因表达,影响胆汁酸组的整体轮廓特征。
罗格列酮;高胆固醇血症;胆汁酸;胆汁酸通路关键基因
罗格列酮是过氧化物酶体增殖体激活体受体(PPAR)的激动剂,除通过胰岛素增敏调控血糖外,还对PPAR介导的胆固醇代谢有调控作用[1,2]。目前,关于罗格列酮的药理作用机制仍然不够全面。除PPAR外,其他核受体也可能在其中扮演重要的角色。在胆固醇的调节中,法尼醇X受体(FXR)的作用极其关键[4],研究罗格列酮是否通过FXR通路发挥药效以及如何对该通路进行影响具有重要临床意义。在FXR所调控的下游通路中,胆汁酸代谢最为独特,也与FXR的调控最为相关,经常通过胆汁酸通路被调控程度及胆汁酸组成特征变化来反映FXR生物功能的调控,其中肝脏[5~9]和肠道[10,11]两条途径非常重要。胆汁酸通路除能够灵敏的反映FXR生物功能变化外,其本身与糖脂代谢之间也存在密切关系[12~14]。本研究观察了罗格列酮对小鼠高胆固醇血症的治疗作用、肝脏胆汁酸代谢影响,并探讨其可能机制。
1 材料与方法
1.1 主要试剂及仪器 TRIzol(美国invitrogen公司);DEPC水(北京庄盟国际生物基因科技有限公司);SYBR Green Supermix(美国Bio-Rad公司);逆转录试剂盒(宝日医生物技术有限公司);TE缓冲液(pH 8.0,北京索莱宝科技有限公司);胆汁酸对照品(美国Sigma-Aldrich公司);实时定量PCR引物根据UCSC小鼠mm10基因组,采用Primer5进行设计后,由上海捷瑞生物工程有限公司代为合成;高脂饲料(美国Research Diet公司);乙腈、甲醇等色谱纯试剂(美国Tedia公司);色谱纯乙酸铵(美国Thermo Fisher Scientific公司);罗格列酮钠片(太极集团四川太极制药有限公司);其他试剂均为分析纯(成都市科龙化工试剂厂)。逆转录用PCR仪(德国Eppendorf公司);实时定量PCR仪(美国Bio-rad公司);NANO DROP 2000微量分光光度计(美国Thermo公司);Y10型超细匀浆机(上海翼控机电有限公司);DN-36A型水浴氮吹仪(上海乔枫实业有限公司);Agilent 6420超高效液相色谱联用三重四级杆质谱(美国安捷伦科技有限公司);-80 ℃超低温冰箱(海尔公司)。
1.2 实验动物分组及罗格列酮干预 C57/BL小鼠24只,自由摄入高脂饲料16周,制备高胆固醇血症模型。造模成功后,随机分为两组,干预组灌胃罗格列酮(1.2 mg/kg)羧甲基纤维素钠混悬液,1次/d,持续22周;模型组灌胃等体积的空白羧甲基纤维素钠溶液;上述两组小鼠造模成功直至给药结束期间,持续给予高脂饲料。另取12只C57/BL小鼠作对照(正常组),始终给予普通饲料,每日灌胃等体积空白羧甲基纤维素钠溶液。动物实验结束后,以7%水合氯醛(0.05 mL/10 g)麻醉,摘眼球取血,离心,分离血清待测。取肝脏,部分组织10%甲醛溶液固定,其余置于液氮速冻后-80 ℃保存。取十二指肠段,生理盐水清洗内容物,置液氮速冻后-80 ℃保存。
1.3 组织病理学检查 将肝脏组织以10%甲醛溶液固定24 h,从低至高浓度的酒精脱水后,二甲苯处理,蜡块包埋,切6~8 μm薄片,贴片,45 ℃烘干。室温平衡15 min后,蒸馏水水化,苏木素溶液染色4 min,流水冲洗数秒,盐酸乙醇中分色数秒,流水冲洗至细胞核呈现蓝色,入伊红溶液中染色2 min,水洗2~3 min。染色后的切片先后经80%和90%酒精中脱水数秒,无水乙醇中先后脱水各1 min,封片,显微镜下观察。
1.4 血生化指标检测 血糖采用罗氏公司血糖仪进行测定。血清总胆固醇(TC)、低密度脂蛋白胆固醇(LDL-C)、谷丙转氨酶(ALT)、谷草转氨酶(AST)等指标采用南京建成生物工程研究所试剂盒进行测定。结果以相对于正常组的倍数变化表示(正常组TC、LDL、血糖、AST、ALT设定值分别为1.00±0.03、1.00±0.15、1.00±0.03、1.00±0.19、1.00±0.17)。
1.5 肝脏胆汁酸分析 取肝脏组织100 mg,加甲醇(1 mg/3 μL)匀浆,12 000×g低温下离心10 min,取上清250 μL氮气吹干,残渣用100 μL的50%甲醇复溶。复溶液相同参数再次离心后,取上清进行LC-MS分析。采用Agilent 6420超高效液相色谱联用三重四级杆质谱系统,固定相Waters BEH C18色谱柱(1.7 μm,2.1×100 mm),以乙腈及乙酸铵溶液为流动相,梯度洗脱参考文献[15]。质谱参数为Gas Temp:326 ℃,Gas Flow:12 L/min,Nebulizer:55 psi,Capillary:3 500 V。质量分析在负离子模式下采用选择离子检测方式完成。
1.6 胆汁酸通路关键基因分析 分别取肝脏、肠道组织各30 mg,置1 mL TRIzol溶液中冰上匀浆,匀浆液以12 000×g低温下离心15 min,取分离上清,与200 μL氯仿混合,反复振摇充分萃取,相同参数再次离心,转移上清与500 μL异丙醇混合,充分混匀,相同参数再次离心,弃去上清。75%乙醇(DEPC水配制,1 000 μL)洗涤沉淀,7 500×g低温下离心15 min,再次弃去上清。沉淀常温放置,待挥干后加入DEPC水复溶,用超微量分光光度计检测RNA浓度及质量。1 μg RNA采用随机引物及逆转录酶逆转录为cDNA后,用于实时定量PCR(RT-PCR)分析。结果以相对于正常组的倍数变化表示(正常组胆汁酸通路关键基因FXR、Fgf15、Fgfr4、JNK1、JNK2、Erk1、Erk2、CYP7A1、CYP8B1、FXR、SHP、Lrh1、Hnf4α设定值分别为1.00±0.07、1.00±0.11、1.00±0.10、1.00±0.14、1.00±0.13、1.00±0.12、1.00±0.12、1.00±0.18、1.00±0.15、1.00±0.06、1.00±0.16、1.00±0.07、1.00±0.08)。

2 结果
2.1 各组组织病理学检查结果比较 组织病理学检查发现,模型组肝细胞中充斥大量脂肪泡,干预组细胞内脂肪泡与正常组相近(图1)。
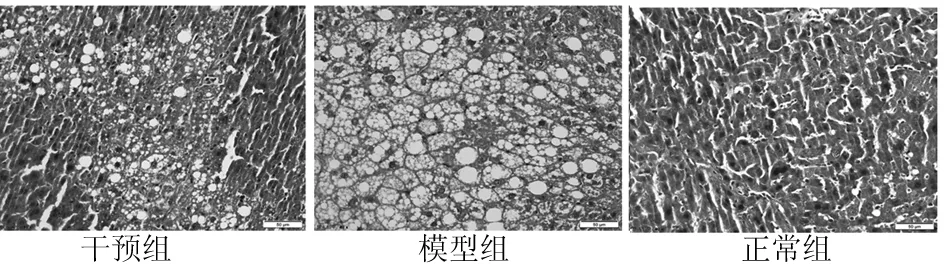
图1 各组组织病理学检查结果(×40)
2.2 模型组、干预组血生化指标相较于正常组的倍数比较 模型组TC、LDL、血糖、AST、ALT相对于正常组的倍数分别为2.13±0.22、1.91±0.38、1.17±0.05、1.61±0.16、3.05±1.11,干预组相对于正常组的倍数分别为1.15±0.09、0.93±0.20、0.95±0.03、0.99±0.28、16.64±3.60,与模型组比较,干预组TC、LDL、血糖水平降低,ALT水平升高(P均<0.05)。
2.3 各组胆汁酸分析结果比较 本研究中采用LC-MS检测了14种胆汁酸(图2),包括游离型胆汁酸5种、甘氨酸(G)结合胆汁酸3种、牛磺酸(T)结合胆汁酸6种,基本覆盖了肝脏中大部分高丰度胆汁酸。为了快速找到这些胆汁酸中,与造模及治疗意义相关的胆汁酸,本研究采用主成分析法对胆固醇及血糖变化典型的小鼠(每组5只)进行了分析,结果表明3组小鼠在不同的维度可以得到显著区分(见不同虚线椭圆,图3A)。通过载荷图分析可知,造模后以及罗格列酮治疗后变化最显著的胆汁酸分子分别是牛碘酸胆酸(TCA)和脱氧胆酸(DCA)(图3B)。为验证PCA的结果,将TCA和DCA按照传统统计方法进行统计,结果表明和主成分分的结果一致。TCA在造模后显著下降,但是罗格列酮没有能逆转其下降的趋势(表1)。DCA在造模后没有太大变化,但是罗格列酮给药后,DCA急剧上升,远远超过对照组正常水平(表1)。
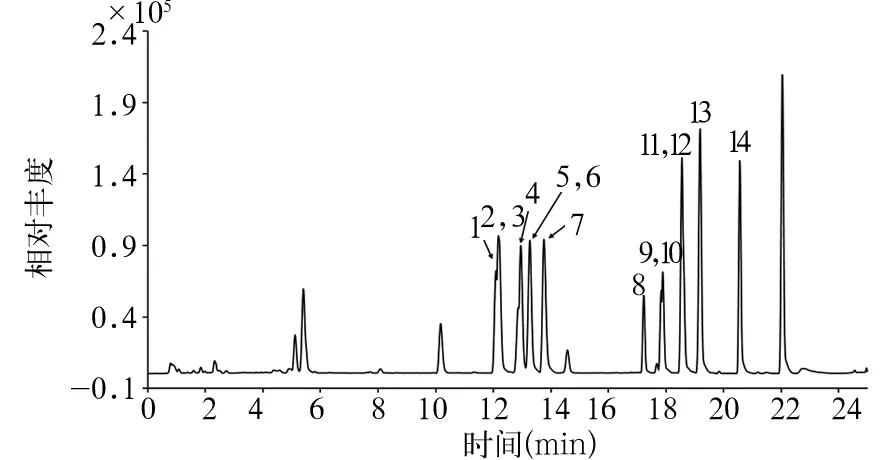
注:1为牛磺酸熊去氧胆酸,2为甘氨酸胆酸,3为熊去氧胆酸,4为牛磺酸猪去氧胆酸,5为胆酸,6为猪去氧胆酸,7为牛磺酸胆酸,8为甘氨酸鹅去氧胆酸,9为牛磺酸鹅去氧胆酸,10为甘氨酸脱氧胆酸,11为牛磺酸脱氧胆酸,12为鹅去氧胆酸,13为脱氧胆酸,14为牛磺酸石胆酸。
图2 胆汁酸总离子流图
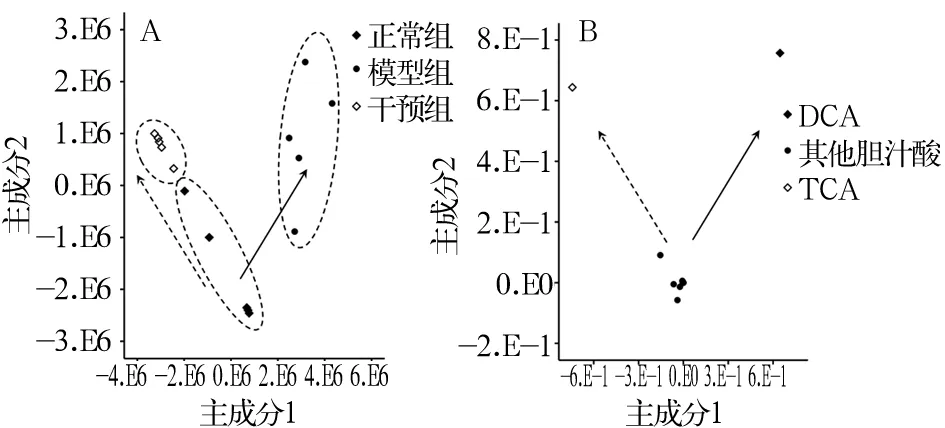
注:A为主成分分析得分图,B为主成分分析载荷图。

图3 肝脏胆汁酸主成分
注:与正常组比较,△P<0.05;与模型组比较,*P<0.05。
2.4 模型组、干预组胆汁酸通路关键基因相较于正常组的倍数比较 模型组FXR、Fgf15、Fgfr4、JNK1、JNK2、Erk1、Erk2、CYP7A1、CYP8B1、FXR、SHP、Lrh1、Hnf4α基因表达相对于正常组的倍数分别为0.40±0.05、0.47±0.07、0.52±0.07、0.32±0.07、0.47±0.07、0.40±0.05、0.43±0.06、0.18±0.06、0.22±0.07、0.46±0.08、0.44±0.07、0.42±0.05、0.48±0.08,干预组相对于正常组的倍数分别为0.53±0.04、0.47±0.08、0.59±0.06、0.67±0.14、0.81±0.14、0.51±0.05、1.08±0.15、1.09±0.18、0.28±0.06、1.06±0.10、0.20±0.04、0.56±0.12、0.42±0.03,与模型组比较,干预组JNK1、Erk2、CYP7A1、FXR表达升高,SHP表达降低(P均<0.05)。
3 讨论
本研究显示,罗格列酮对高脂饮食诱导的高胆固醇有显著治疗作用。通过对胆汁酸通路关键基因的分析及胆汁酸分子的定量分析,发现其治疗作用与胆汁酸通路非常相关。Cyp7a1和Cyp8b1是从胆固醇合成胆汁酸的关键酶[16],高脂饮食压力下,两个酶的基因表达都严重下调,这种下调势势必造成胆固醇代谢受阻,进而出现高胆固醇症状。由于胆固醇代谢和血糖代谢非常相关,同时也造成了血糖的显著升高。罗格列酮给药后,Cyp7a1的表达被回调。由于胆汁酸是胆固醇的主要代谢途径[12],这就为降低体内胆固醇的水平创造了条件。LC-MS分析发现,罗格列酮给药后胆汁酸中的DCA分子有70余倍的上升,与Cyp7a1的变化相关性明显,证明了本文的推断。
除直接参与胆汁酸合成的关键酶Cyp7a1和Cyp8b1,胆汁酸调控的肝脏途径的相关基因也在罗格列酮的作用下发生了相应的显著变化。首先是肝脏FXR,其为胆汁酸调控的核心成员[17,18],本研究显示在高脂饮食压力下被显著抑制,这与文献[19]报道非常一致。FXR下游通路中SHP、Lrh1、Hnf4α也被抑制,理论上这一通路的抑制原本会造成Cyp7a1和Cyp8b1的上升[7~9],但是本文和文献均观察到与之对应的是Cyp7a1、Cyp8b1下调。这应该是机体在感应到胆汁酸合成受阻后发生的负反馈调节作用,只是由于持续的高脂饮食压力使得这种负反馈调节作用在没有药物干预的情况下难以发挥作用。罗格列酮给药后,通过进一步的下调SHP,强化了对Cyp7a1的上调,故最终完成了通过打通胆固醇向胆汁酸合成而恢复糖脂代谢稳定的任务。本研究还显示,罗格列酮给药后,在SHP、Cyp7a1之间的Lrh1、Hnf4α表达并没有发生变化。据报道,SHP可以在蛋白水平与Lrh1、Hnf4α发生杂合[6,7],进而抑制后两者的功能。因此,尽管转录水平没有发生变化,SHP仍然可以在蛋白相互作用的水平抑制Lrh1以及Hnf4α,进而实现对Cyp7a1的上调。
在胆汁酸调控的肠道途径,胆汁酸进入肠道后,刺激肠道FXR进而诱导其下游因子肠道Fgf15及肝脏Fgfr4、JNK、ERK等表达[10,11]。本研究显示,肠道FXR、Fgf15以及肠道Fgf15循环进入肝脏后的下游目标JNK、ERK都在高脂饮食压力下全面下调。这有可能是由于高脂饮食压力下胆固醇代谢受阻导致排入肠道胆汁酸减少,使肠道FXR缺乏刺激所致。罗格列酮给药后,除JNK1和ERK2外,肠道FXR、Fgf15及肝脏Fgfr4、JNK2、ERK1等仍保持下调趋势,为Cyp7a1的上调创造了条件。JNK1和ERK2的显著上调可能是DCA发生异常升高后机体的负反馈调节措施。
除药效外,本研究还观察到罗格列酮给药后ALT的异常升高。这表明罗格列酮给药后产生了明显的肝脏功能紊乱,至少有一定程度肝损伤存在。罗格列酮给药的肝损伤问题已经引起关注[20,21],但是其产生肝脏毒性的机制一直不明确。本文在利用LC-MS技术对胆汁酸进行分析后,发现胆汁酸中的DCA分子在干预组发生了异常的升高,其升高值是正常组的70余倍。DCA是胆汁酸分子中相对疏水性较强的分子之一,其肝脏毒性已经广为报道[22,23]。因此,本研究中观察到的DCA的异常很可能是罗格列酮造成肝损伤的重要机制,值得进一步深入研究。
[1] Mayerson AB, Hundal RS, Dufour S, et al. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes[J]. Diabetes, 2002,51(3):797-802.
[2] Gruszka A, Kunert-Radek J, Pawlikowski M. Rosiglitazone, PPAR-gamma receptor ligand, decreases the viability of rat prolactin-secreting pituitary tumor cells in vitro[J]. Neuro Endocrinol Lett, 2005,26(1):51-54.
[3] Graham DJ, Ouellet-Hellstrom R, MaCurdy TE, et al. Risk of acute myocardial infarction, stroke, heart failure, and death in elderly Medicare patients treated with rosiglitazone or pioglitazone [J]. JAMA, 2010,304(4):411-418.
[4] Thomas AM, Hart SN, Kong B, et al. Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine[J]. Hepatology, 2010,51(4):1410-1419.
[5] Matsubara T, Li F, Gonzalez FJ. FXR signaling in the enterohepatic system[J]. Mol Cell Endocrinol, 2013,368(1-2):17-29.
[6] Lee YK, Dell H, Dowhan DH, et al. The orphan nuclear receptor SHP inhibits hepatocyte nuclear factor 4 and retinoid X receptor transactivation: two mechanisms for repression[J]. Mol Cell Biol, 2000,20(1):187-195.
[7] Goodwin B, Jones SA, Price RR, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis [J]. Mol Cell, 2000,6(3):517-526.
[8] Kir S, Zhang Y, Gerard RD, et al. Nuclear receptors HNF4alpha and LRH-1 cooperate in regulating Cyp7a1 in vivo[J]. J Biol Chem, 2012,287(49):41334-41341.
[9] Inoue Y, Yu AM, Yim SH, et al. Regulation of bile acid biosynthesis by hepatocyte nuclear factor 4alpha [J]. J Lipid Res, 2006,47(1):215-227.
[10] Stroeve JH, Brufau G, Stellaard F, et al. Intestinal FXR-mediated FGF15 production contributes to diurnal control of hepatic bile acid synthesis in mice[J]. Lab Invest, 2010,90(10):1457-1467.
[11] Kong B, Wang L, Chiang JY, et al. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice[J]. Hepatology, 2012,56(3):1034-1043.
[12] Russell DW. The enzymes, regulation, and genetics of bile acid synthesis[J]. Annu Rev Biochem, 2003,72:137-174.
[13] Islam KB, Fukiya S, Hagio M, et al. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats [J]. Gastroenterology, 2011,141(5):1773-1781.
[14] Kobayashi M, Ikegami H, Fujisawa T, et al. Prevention and treatment of obesity, insulin resistance, and diabetes by bile acid-binding resin[J]. Diabetes, 2007,56(1):239-247.
[15] Zhang Y, Klaassen CD. Effects of feeding bile acids and a bile acid sequestrant on hepatic bile acid composition in mice [J]. J Lipid Res, 2010,51(11):3230-3242.
[16] Chiang JY. Bile acids: regulation of synthesis[J]. J Lipid Res, 2009,50(10):1955-1966.
[17] Lee FY, Lee H, Hubbert ML, et al. FXR, a multipurpose nuclear receptor[J]. Trends Biochem Sci, 2006,31(10):572-580.
[18] Westin S, Heyman RA, Martin R. FXR, a therapeutic target for bile acid and lipid disorders[J]. Mini Rev Med Chem, 2005,5(8):719-727.
[19] He X, Zheng N, He J, et al. Gut microbiota modulation attenuated the hypolipidemic effect of simvastatin in high-fat/cholesterol-diet fed mice[J]. J Proteome Res, 2017,16(5):1900-1910.
[20] Al-Salman J, Arjomand H, Kemp DG, et al. Hepatocellular injury in a patient receiving rosiglitazone. A case report[J]. Ann Intern Med, 2000,132(2):121-124.
[21] Bissell DM, Gores GJ, Laskin DL, et al. Drug-induced liver injury: mechanisms and test systems [J]. Hepatology, 2001,33(4):1009-1013.
[22] Delzenne NM, Calderon PB, Taper HS, et al. Comparative hepatotoxicity of cholic acid, deoxycholic acid and lithocholic acid in the rat: in vivo and in vitro studies [J]. Toxicol Lett, 1992,61(2-3):291-304.
[23] Hassoun EA, Cearfoss J, Spildener J. Dichloroacetate- and trichloroacetate-induced oxidative stress in the hepatic tissues of mice after long-term exposure[J]. J Appl Toxicol, 2010,30(5):450-456.
国家自然科学基金资助项目(81402985,81560673,81660685);贵州省科技重大专项(黔科合重大专项字[2015]6010);贵州省科学技术基金(黔科合J字[2015]2158号,黔科合JZ字[2015]2010号);贵州省出国留学人员择优资助计划(黔人项目资助合同(2015)03号);上海市复方中药重点实验室开放基金(14DZ2271000);国家级/省级大学生创新创业项目(201510661002);遵义医学院大学生创新创业项目(遵医[2015]5046);贵州省药剂学研究生卓越人才培养计划(黔教研合ZYRC字[2014]019)。
鲁艳柳(E-mail: yanliu.lu@foxmail.com、王玉和(E-mail: wangyuhe11@163.com)
10.3969/j.issn.1002-266X.2017.31.009
R966
A
1002-266X(2017)31-0032-04
2017-03-13)
猜你喜欢
英语文摘(2021年11期)2021-12-31 03:25:24
理化检验-化学分册(2021年10期)2021-11-29 14:50:42
课外语文·中(2019年10期)2019-11-28 11:43:58
金山(2016年9期)2016-10-12 14:22:23
天津医科大学学报(2015年3期)2015-06-05 12:21:49
天津医科大学学报(2015年3期)2015-06-05 12:21:49
中国当代医药(2015年9期)2015-03-01 02:02:12
现代检验医学杂志(2015年2期)2015-02-06 02:00:50
中成药(2014年4期)2014-04-01 08:43:42
湖北科技学院学报(医学版)(2014年3期)2014-02-28 19:42:51
