
小菜蛾传染性软腐病病毒的全基因组序列及其进化分析
2017-08-02 01:39陈大嵩戴建青韩诗畴
环境昆虫学报 2017年3期
陈大嵩,戴建青,韩诗畴
小菜蛾传染性软腐病病毒的全基因组序列及其进化分析
陈大嵩,戴建青*,韩诗畴
(广东省生物资源应用研究所,广东省动物保护与资源利用重点实验室,广东省野生动物保护与利用公共实验室,广州 510260)
传染性软腐病病毒(Iflavirus)属于小核糖核酸病毒目,是一类单股正链RNA病毒,其家族成员正在不断扩大。Iflavirus的寄主包括鳞翅目、膜翅目和半翅目的昆虫以及瓦螨,因其对一些重要的农业害虫产生致病性,因此是重要的潜在生防病毒种类之一。本研究利用小菜蛾转录组测序数据拼接出一条Iflavirus病毒基因组序列。通过查找ORF和序列相似性比对发现此基因组可编码一条完整的病毒多聚蛋白并与其他Iflavirus病毒同源,因此将这一新病毒命名为小菜蛾传染性软腐病病毒(Diamondback moth iflavirus),英文简称DBMIV。通过多聚蛋白序列与RNA聚合酶序列构建的进化树表明DBMIV与PNV、EOPLV和SEIV2进化关系最近。最后,利用RT-PCR扩增出DBMIV的外壳蛋白区域验证了新命名病毒在田间小菜蛾种群的感染。DBMIV的鉴定与命名为利用高通量测序数据鉴定病毒新种提供参考,为利用病毒生防重要蔬菜害虫小菜蛾提供研究思路。
小菜蛾;传染性软腐病病毒;基因组;系统进化
病毒存在于自然界的各个角落,几乎感染任何物种(Dinsdaleetal.,2008)。以昆虫为寄主的RNA病毒种类包括传染性软腐病病毒科Iflaviridae、双顺反子病毒科Dicistroviridae、诺达病毒科Nodaviridae以及质型多角体病毒属(Reoviridae:Cypovirus)(Asgari & Johnson,2010)。相对于其他昆虫RNA病毒,传染性软腐病病毒和双顺反子病毒的寄主包括蜜蜂、家蚕以及农业害虫等重要昆虫,所以被研究的更加广泛。传染性软腐病病毒与双顺反子病毒同属于小核糖核酸病毒目(Picornavirales)。截至目前为止(2017年1月),已经报道了几十种传染性软腐病病毒种类,共29条病毒基因组序列(表1),寄主包括鳞翅目Lepidoptera、膜翅目Hymenoptera和半翅目Hemiptera的多种昆虫以及瓦螨Varroadestructor等节肢动物,是最重要的昆虫RNA病毒种类之一。
传染性软腐病病毒起初在蜜蜂(Baileyetal.,1964)、家蚕(Kawaseetal.,1980)等经济昆虫中被分离并鉴定出来,其病毒颗粒是与其他小核糖核酸病毒类似的正二十面体(van Oers,2010),其基因组是单股正链RNA(Baileyetal.,1964),基因组RNA的3端具有多聚腺苷酸(Poly(A))结构(Ongusetal.,2004)。除去Poly(A)结构,基因组长度大约在9000-10000 bp(表1)。传染性软腐病病毒基因组包含一个长ORF,编码一条多聚蛋白质,如同其他小核糖核酸病毒一样没有亚基因组(Wuetal.,2002)。多聚蛋白由N端到C端为前导肽(Leader peptide)、4个外壳蛋白、解旋酶、蛋白酶和RNA聚合酶。多聚蛋白被翻译后会被蛋白酶分裂成成熟的、具备各自相应功能的蛋白质。传染性软腐病病毒与双顺反子病毒同属于小核糖核酸病毒种类,但他们的基因组相差很大。双顺反子病毒拥有两个ORF,ORF之间有一个基因间隔区且行使内部核糖体进入位点(Internal ribosome entry site,IRES)的功能,靠近5端的ORF编码结构蛋白而靠近3端的编码非结构蛋白,而传染性软腐病病毒只有一个ORF;传染性软腐病病毒ORF的5端能够编码前导肽,而双顺反子病毒没有编码前导肽。真核生物进行蛋白翻译时,核糖体需要识别mRNA的5端帽子(cap)结构(Pestovaetal.,2001),而传染性软腐病病毒与双顺反子病毒RNA的5端没有帽子结构,但其5端非编码区(5′UTR)都存在内部核糖体进入位点,允许病毒RNA在5端没有帽子结构的情况下招募核糖体并翻译多聚蛋白(Doudna & Sarnow,2007)。
传染性软腐病病毒可引起寄主腹泻、发育畸形以及致死等多种病症。例如囊仔病(Sacbrood)能够引起幼蜂外壳下液体的积聚形成液囊,最终引起幼蜂的死亡(Baileyetal.,1964)。残翅病毒(Deformed wing virus)是另一种感染蜜蜂的病毒,其能够引起翅的发育畸形并导致蜜蜂的致残(Yue & Genersch,2005)。与残翅病毒遗传进化关系密切的kakugo病毒则能够入侵蜜蜂的脑组织,引起蜜蜂攻击性的增强(Fujiyukietal.,2004)。家蚕传染性软化病毒(Infectious flacherie virus)则能够感染家蚕中肠上皮组织并引起病变,进而引起细菌的二次感染,毙死后尸体软化腐烂(Ayuzawa,1972)。虽然许多传染性软腐病病毒对寄主致病,但是另一些的感染却是无症状的,如褐飞虱蜜露病毒虽然能够在褐飞虱个体间传染,却未观察到明显病症(Murakamietal.,2013a;Murakamietal.,2013b)。
随着高通量测序技术的发展,推进了测序技术在生物学问题中的应用,虽然其在昆虫病毒的研究中才刚刚起步,但是却能够挖掘出丰富的病毒种类(Shietal.,2016)。随着越来越多的病毒种类被高通量测序所鉴定出来,其数量远远大于传统的分离鉴定方法,因此一些学者也提出来制定合适的数据质控流程,把高通量测序所鉴定出来的病毒种类编入国际病毒分类学委员会(International Committee on Taxonomy of Viruses,ICTV)的病毒命名名单中(Simmondsetal.,2017)。在本文中,作者通过拼接小菜蛾转录组测序数据,获得一条具有完整的病毒基因组特征的序列。鉴于其可能来自于未命名的病毒种类,作者将其命名为小菜蛾传染性软腐病病毒(Diamondback moth iflavirus)。对小菜蛾传染性软腐病病毒的命名以及其序列的分析,将为利用高通量数据鉴定昆虫病毒以及其他动物病毒新种提供参考,并且丰富了传染性软腐病病毒家族的病毒种类,为生物防控小菜蛾在经济作物中的种群数量提供新思路。
1 材料与方法
1.1 DBMIV病毒基因组的序列分析
本实验室所获得的小菜蛾转录组原始序列经过过滤后,利用Trinity软件拼接出小菜蛾传染性软腐病病毒(Diamondback moth iflavirus,DBMIV)的基因组序列(Haasetal.,2013)。ORF开放阅读框利用在线分析工具ORF finder(www.ncbi.nlm.nih.gov/orffinder)查询,遗传密码子选择通用密码子(Standard),并查找只以“ATG”起始的阅读框。利用MEGA7软件分析核酸组成、氨基酸组成以及同义密码子频率(Kumaretal.,2016)。利用Blastn进行核酸序列与NT数据库的序列相似性比对,利用Blastx进行多聚蛋白与NR数据库的序列相似性比对(Camachoetal.,2009)。
1.2 软腐病病毒系统发育关系的重建
传染性软腐病病毒科与双顺反子病毒科的氨基酸序列从GenBank获得,其GenBank登录号见表1。利用传染性软腐病病毒科的多聚蛋白序列在MEGA软件上以Muscle算法进行多序列比对,比对参数选择默认值,最终序列矩阵为4623 aa。利用传染性软腐病病毒科的多聚蛋白与双顺反子病毒科的第一段含有非结构蛋白的ORF以Muscle算法进行多序列比对,比对参数选择默认值,截取总长1979 aa的RNA聚合酶基因序列矩阵进行序列进化分析。为了确定DBMIV为病毒新种,利用MEGA软件以p-distance为模型计算传染性软腐病病毒科多聚蛋白序列之间的遗传距离,自展(Bootstrap)1000次计算标准差。利用MEGA软件计算最大似然法替代模型,根据贝叶斯信息评判标准(Bayesian Information Criterion,BIC)选择出最优替代模型为LG+G+I+F(次优替代模型为LG+G+I)。利用PhyML 3.1软件构建最大似然法进化树(Guindonetal.,2010),进化模型选择LG,Proportion of invariable sites 选择estimated (+I),Gamma distribution parameter 选择estimated (+G),Amino acid frequencies选择empirical(+F),置信值利用aLRT算法计算(Anisimova & Gascuel,2006)。生成的进化树文件用MEGA软件修改并输出图片。利用MrBayes 3.2.5软件构建贝叶斯进化树(Ronquistetal.,2012),氨基酸模型选择LG,取样频率为10,每100代计算一次分裂频率平均标准差,共计算106代后检查标准差的大小并利用Tracer 1.6软件(tree.bio.ed.ac.uk/software/tracer/)对马尔科夫链的蒙特卡洛循环(MCMC)进行分析与检验,如计算不充足则继续追加计算代数,最后舍弃前25%的老化样本构建进化树与计算后验概率。利用TreeGraph 2(Stöver & Müller,2010)导出含有后验概率的Newick进化树文件利用MEGA软件修改并输出图片,最后利用Adobe Illustrator软件更换后验概率数值的格式。
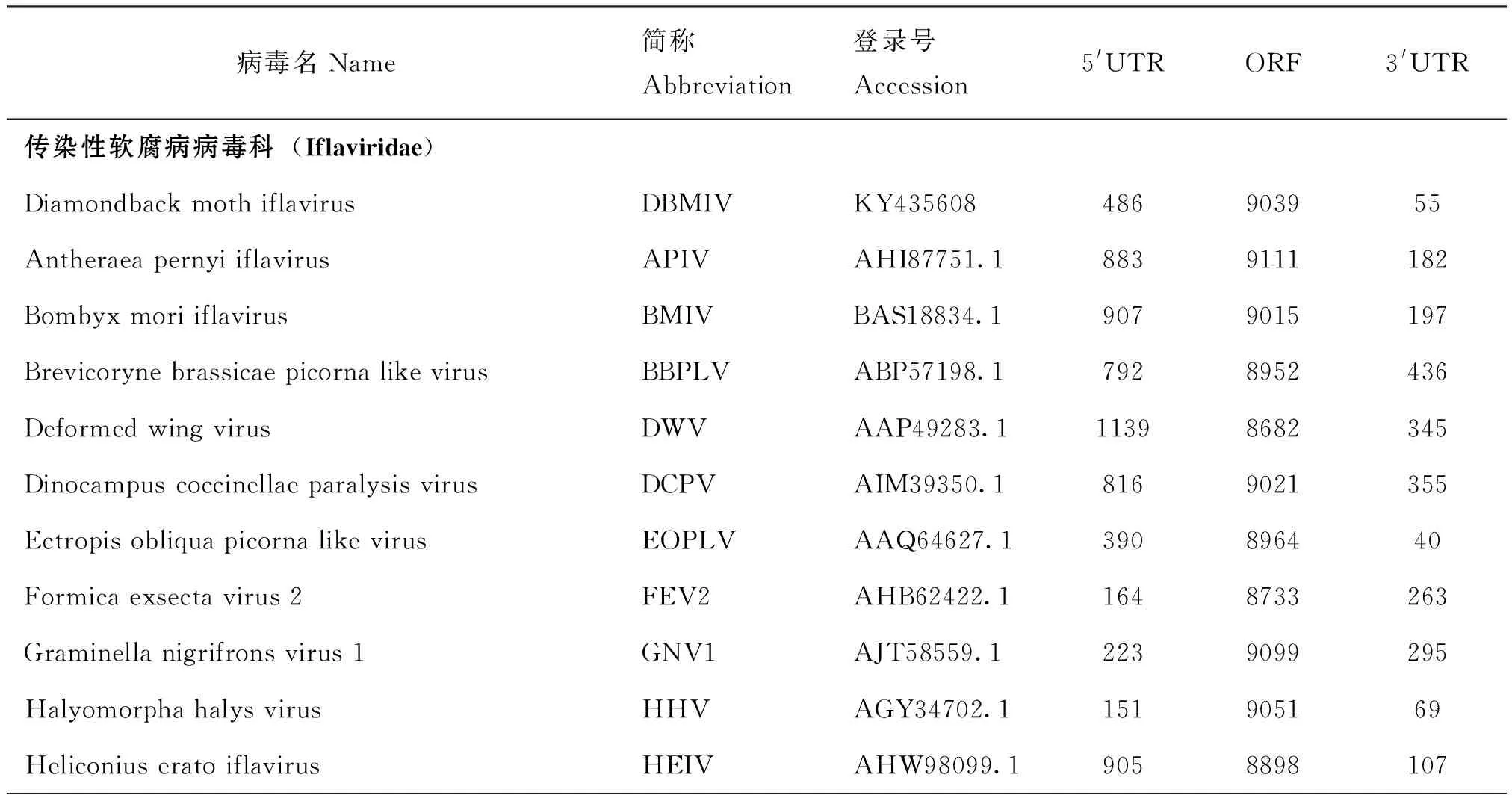
表 1 传染性软腐病与双顺反子病毒科种类的统计Table 1 The summary of the Iflaviridae and Dicistroviridae virus species
续上表
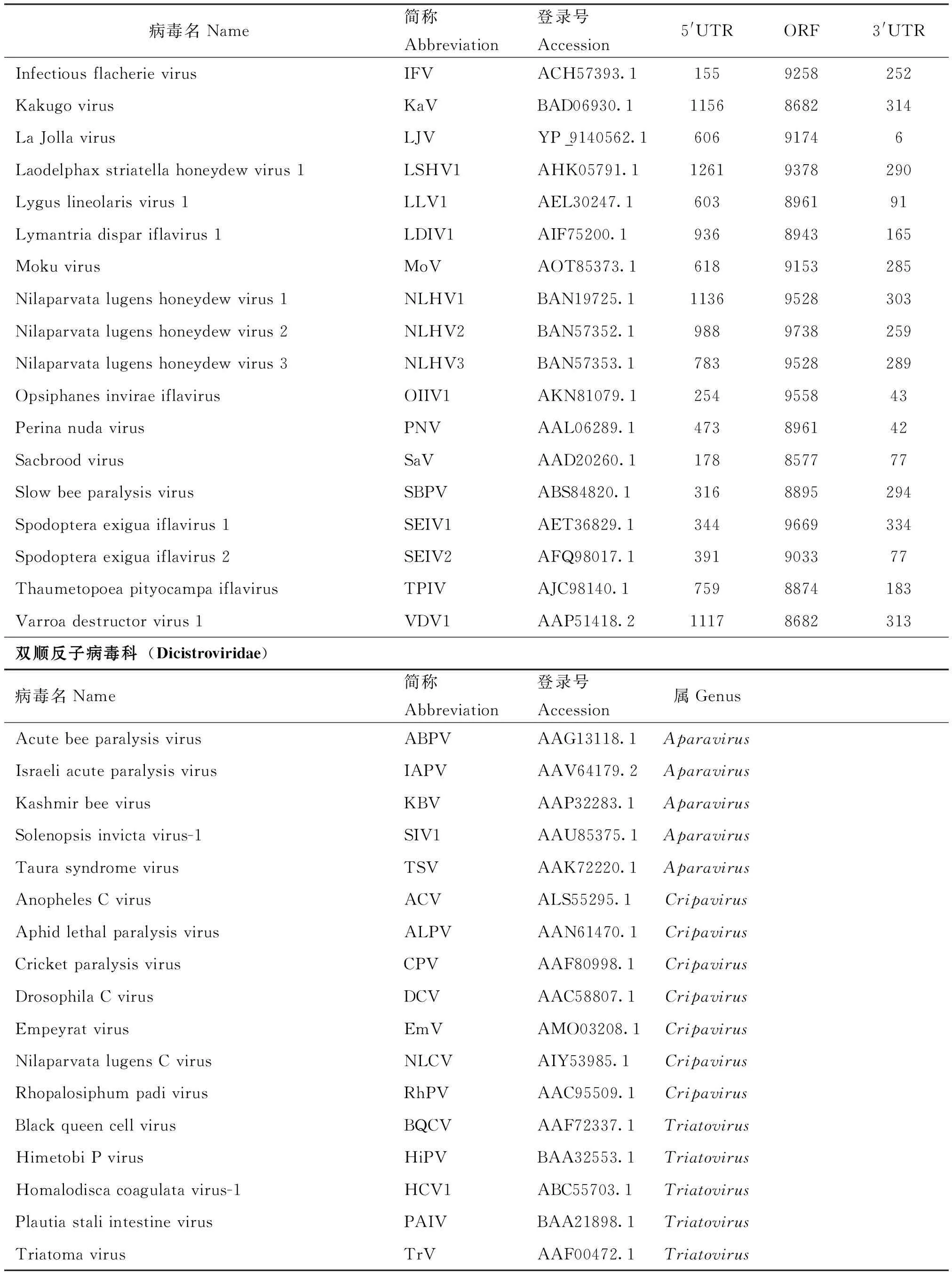
病毒名Name简称Abbreviation登录号Accession5'UTRORF3'UTRInfectiousflacherievirusIFVACH57393.11559258252KakugovirusKaVBAD06930.111568682314LaJollavirusLJVYP_9140562.160691746Laodelphaxstriatellahoneydewvirus1LSHV1AHK05791.112619378290Lyguslineolarisvirus1LLV1AEL30247.1603896191Lymantriadispariflavirus1LDIV1AIF75200.19368943165MokuvirusMoVAOT85373.16189153285Nilaparvatalugenshoneydewvirus1NLHV1BAN19725.111369528303Nilaparvatalugenshoneydewvirus2NLHV2BAN57352.19889738259Nilaparvatalugenshoneydewvirus3NLHV3BAN57353.17839528289OpsiphanesinviraeiflavirusOIIV1AKN81079.1254955843PerinanudavirusPNVAAL06289.1473896142SacbroodvirusSaVAAD20260.1178857777SlowbeeparalysisvirusSBPVABS84820.13168895294Spodopteraexiguaiflavirus1SEIV1AET36829.13449669334Spodopteraexiguaiflavirus2SEIV2AFQ98017.1391903377ThaumetopoeapityocampaiflavirusTPIVAJC98140.17598874183Varroadestructorvirus1VDV1AAP51418.211178682313双顺反子病毒科(Dicistroviridae)病毒名Name简称Abbreviation登录号Accession属GenusAcutebeeparalysisvirusABPVAAG13118.1AparavirusIsraeliacuteparalysisvirusIAPVAAV64179.2AparavirusKashmirbeevirusKBVAAP32283.1AparavirusSolenopsisinvictavirus-1SIV1AAU85375.1AparavirusTaurasyndromevirusTSVAAK72220.1AparavirusAnophelesCvirusACVALS55295.1CripavirusAphidlethalparalysisvirusALPVAAN61470.1CripavirusCricketparalysisvirusCPVAAF80998.1CripavirusDrosophilaCvirusDCVAAC58807.1CripavirusEmpeyratvirusEmVAMO03208.1CripavirusNilaparvatalugensCvirusNLCVAIY53985.1CripavirusRhopalosiphumpadivirusRhPVAAC95509.1CripavirusBlackqueencellvirusBQCVAAF72337.1TriatovirusHimetobiPvirusHiPVBAA32553.1TriatovirusHomalodiscacoagulatavirus-1HCV1ABC55703.1TriatovirusPlautiastaliintestinevirusPAIVBAA21898.1TriatovirusTriatomavirusTrVAAF00472.1Triatovirus
注:5′UTR:5端非编码区;ORF:蛋白开放阅读框;3′UTR:3端非编码区。Note: 5′UTR: 5′ untranslated region; ORF: open reading frame; 3′UTR: 3′ untranslated region.
1.3 RT-PCR的引物设计与实验过程
小菜蛾成虫于2017年3月采集于广州市庙贝村(113.4°E,22.8°N),利用RNAiso Plus(TaKaRa)按照实验说明提取总RNA后 ,利用逆转录试剂盒TransScript First-Strand cDNA Synthesis SuperMix(全式金)合成cDNA。利用Oligo 7设计两对DBMIV的RT-PCR扩增引物(DBMIV1和DBMIV2),引物序列为DBMIV1F:5′-CCT GCA CCC GAC ATT TGA ACC-3′,DBMIV1R:5′-AGC TAC CGT CAT AAG CAT ATA GTT CG-3′;DBMIV2F:5′-CGA ACC GCT ATT CAG TGT ATT CTT AGC A-3′,DBMIV2R:5′-ATC AAA CGC ATT GTT GAG GAC-3′,PCR产物长度分别为1035 bp与1150 bp。PCR体系根据EasyTaq DNA Polymerase(全式金)实验说明按比例混合成20 μL后,在PCR扩增仪中运行如下程序:94℃预变性2 min,扩增循环为94℃变性30s,55℃退火30s,72℃延伸90s,运行35个扩增循环后 72℃延伸7 min。PCR产物加入1%浓度琼脂糖凝胶中,在5 V/cm电压下电泳30 min以检测PCR结果,阳性结果用于直接测序。
2 结果与分析
2.1 DBMIV病毒的发现与命名
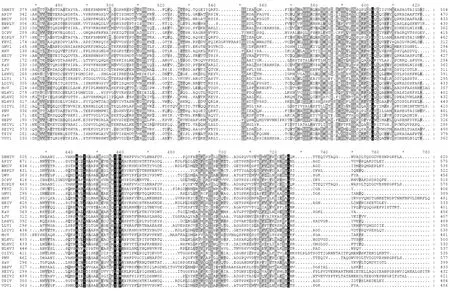
拼接本实验室所获得小菜蛾转录组数据获得一条9580 bp长的序列,利用Blast方法与NCBI的NT数据库比对发现其与一种类小核糖核酸病毒(picorna-like virus)——榕透翅毒蛾病毒(Perina nuda virus,PNV)的基因组序列十分相近,Blastn结果总评分(Total score)为4888 bit,期望值(E value)为0。利用ORF finder查找序列的ORF读码框发现其ORF长9039 bp,可编码一段完整的3012 aa的长肽,5′UTR与3′UTR分别长486 bp和55 bp(图1)。利用Blastp把ORF编码的氨基酸序列与NR数据库做比对,与茶尺蠖类小核糖核酸病毒(Ectropis obliqua picorna-like virus,EOPLV)相似度最高,其次为榕透翅毒蛾病毒,相似区域均占序列总长度(Query cover)的95%。Blastp结果Total score分别为4751 bit与4747 bit,E值均为0,相同的氨基酸位点(Identities)分别为2282和2278,分别占序列比对区域总长的78%和79%,序列比对间隙(Gaps)分别为59和50,分别占序列比对区域总长的2%和1%。氨基酸序列同源序列保守域(Putative conserved domains)包括小核糖核酸病毒衣壳蛋白(Picornavirus capsid protein)、RNA解旋酶(RNA helicase)以及RNA依赖的RNA聚合酶(RNA-dependent RNA polymerase,RdRp)(图2)。鉴于其完整的病毒序列特征,且与传染性软腐病病毒属(Iflavirus)关系相近,其可能是未命名的新病毒,按照病毒命名习惯对其以寄主名加病毒属名的方式命名为小菜蛾传染性软腐病病毒,英文名为Diamondback moth iflavirus,英文缩写为DBMIV。其基因组序列信息提交GenBank保存,登录号(GenBank accession number)为KY435608。
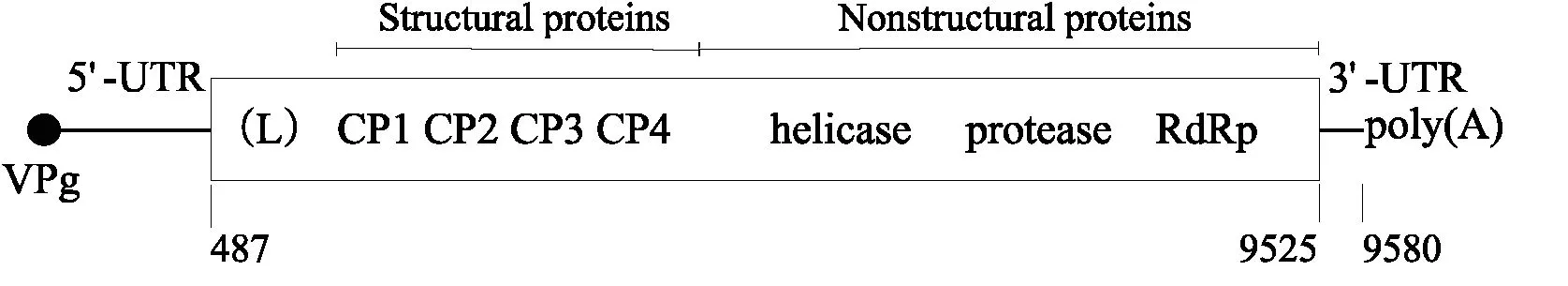
图 1 DBMIV基因组的结构Fig.1 Schematic diagram of the genome organization of DBMIV注:方框代表ORF阅读框,所在碱基位置用数字标记。结构蛋白(外壳蛋白1-4)和非结构性蛋白(解旋酶、蛋白酶以及RNA聚合酶)的大致位置标记在ORF框内,带括号的L代表前导肽,实心圆代表基因组5′端可能连接的病毒基因组结合蛋白(VPg)。Note: The ORF corresponds to the entire open box and the numbers indicate nucleotide positions.The approximate positions of the structural proteins (coat protein 1-4) and nonstructural proteins (helicase, protease and RNA-dependent RNA polymerase, RdRp) are shown in the box.Protein L with parentheses denotes a probable leader peptide.A solid circle represents the genome-linked viral protein (VPg) protein (if presents) that links at the 5′ end of genome.
CP1:
Helicase:

RdRp:
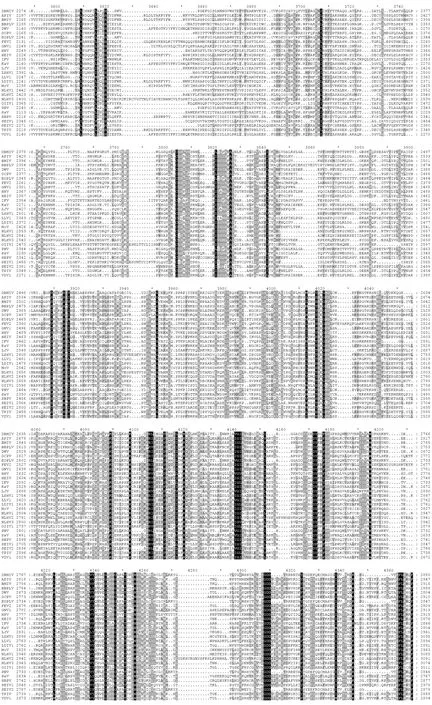
图 2 传染性软腐病病毒蛋白序列的比对。Fig.2 The multiple alignments of the amino acid sequences between the iflaviruses
2.2 DBMIV基因组的特征
DBMIV基因组5端非转录区的GC含量为38%,低于ORF区(46%)以及3端非转录区(44%)(图3上)。ORF区与3端非转录区GC偏倚不同,ORF区GC偏倚为正(G多于C),而3端非转录区GC偏倚为负。密码子的不同位点,受选择压力不同,可能出现不同的核酸组成。ORF区第一位密码子GC含量(51%)明显高于第二位(42%)和第三位(46%)(图3下)。第二位与第三位密码子的主要差别为GC偏倚和AU偏倚不同。总体来说, GC含量的变化主要因为U和G的含量变化造成的。
DBMIV基因组使用最多的几个氨基酸为亮氨酸(L:8.1%)、甘氨酸(G:7.57%)、缬氨酸(V:7.1%)、丙氨酸(A:7.01%)、丝氨酸(S:6.61%)和苏氨酸(T:6.47%),而相对较少出现的氨基酸为组氨酸(H:2.32%)、甲硫氨酸(M:2.29%)、半胱氨酸(C:1.93%)和色氨酸(W:1.43%),但是总体来说并没有极端的使用偏好(图4)。DBMIV基因组使用UAA为终止子,GAU(101)、GAG(98)、AAG(91)、UUU(87)、AAC(76)和AUA(72)为使用最多的密码子,而密码子CGC(19)、CCG(18)、CGU(18)、AGC(15)和CGG(12)使用较少(表2)。
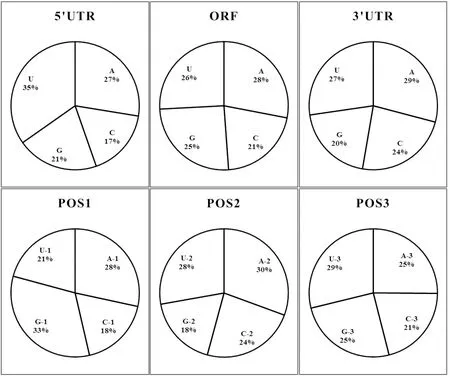
图 3 DBMIV基因组核酸比例分布图Fig.3 Nucleotide frequencies of the DBMIV genome注:由左至右、由上到下分别代表5端非转录区、ORF开放阅读框、3端非转录区、ORF的第1-3个碱基位点的核酸比例。Note: The nucleotide frequencies of 5′UTR, ORF, 3′UTR, the first, the second and the third nucleotide of the ORF were displayed from left to right and from top to bottom.
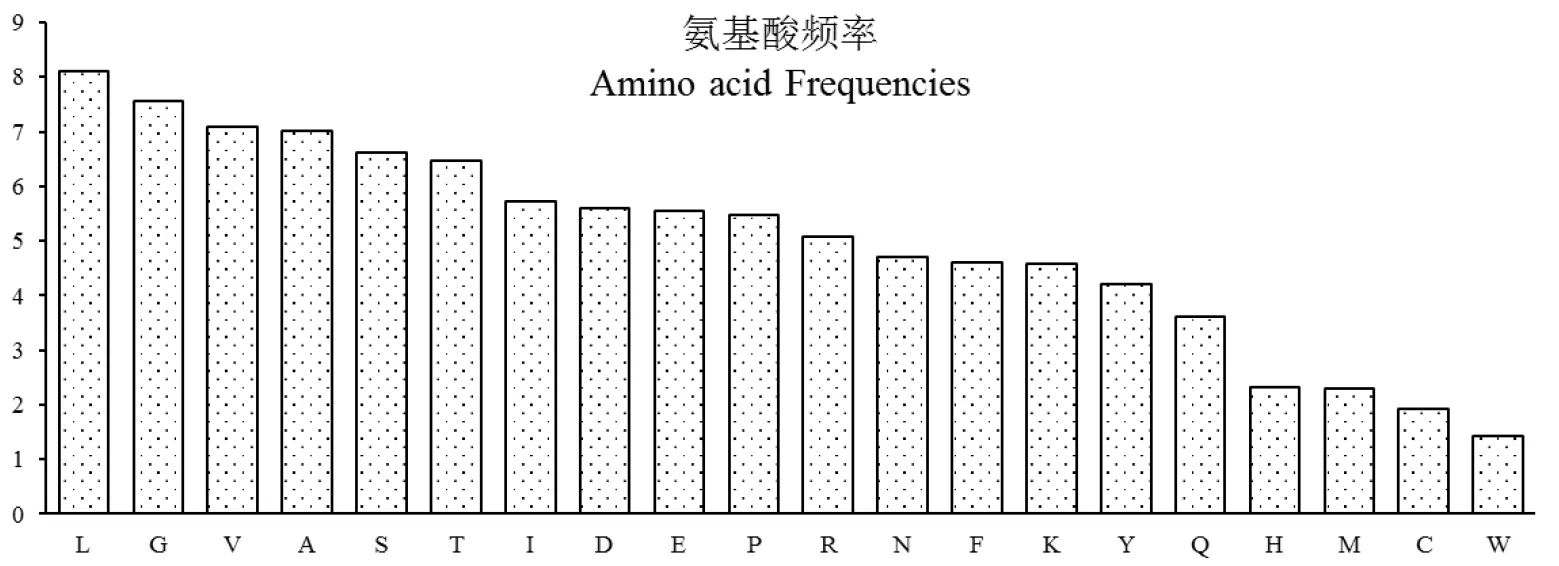
图 4 DBMIV基因组氨基酸频率分布图Fig.4 Amino acid frequencies of the DBMIV genome 注:横坐标代表氨基酸种类,纵坐标代表某个氨基酸的百分比。Note: Amino acids were shown on horizontal axis and the percentages of the amino acid frequencies were displayed as vertical axis.

表 2 DBMIV基因组同义密码子频率表Table 2 Relative synonymous codon usage of the DBMIV genome
注:密码子后的括弧代表氨基酸种类,密码子数量后的括弧代表同义密码子频率。Note: Codons were following up with the amino acids in parentheses.Following the codon frequencies were relative synonymous codon usage given in parentheses.
2.3 DBMIV的系统发育分析
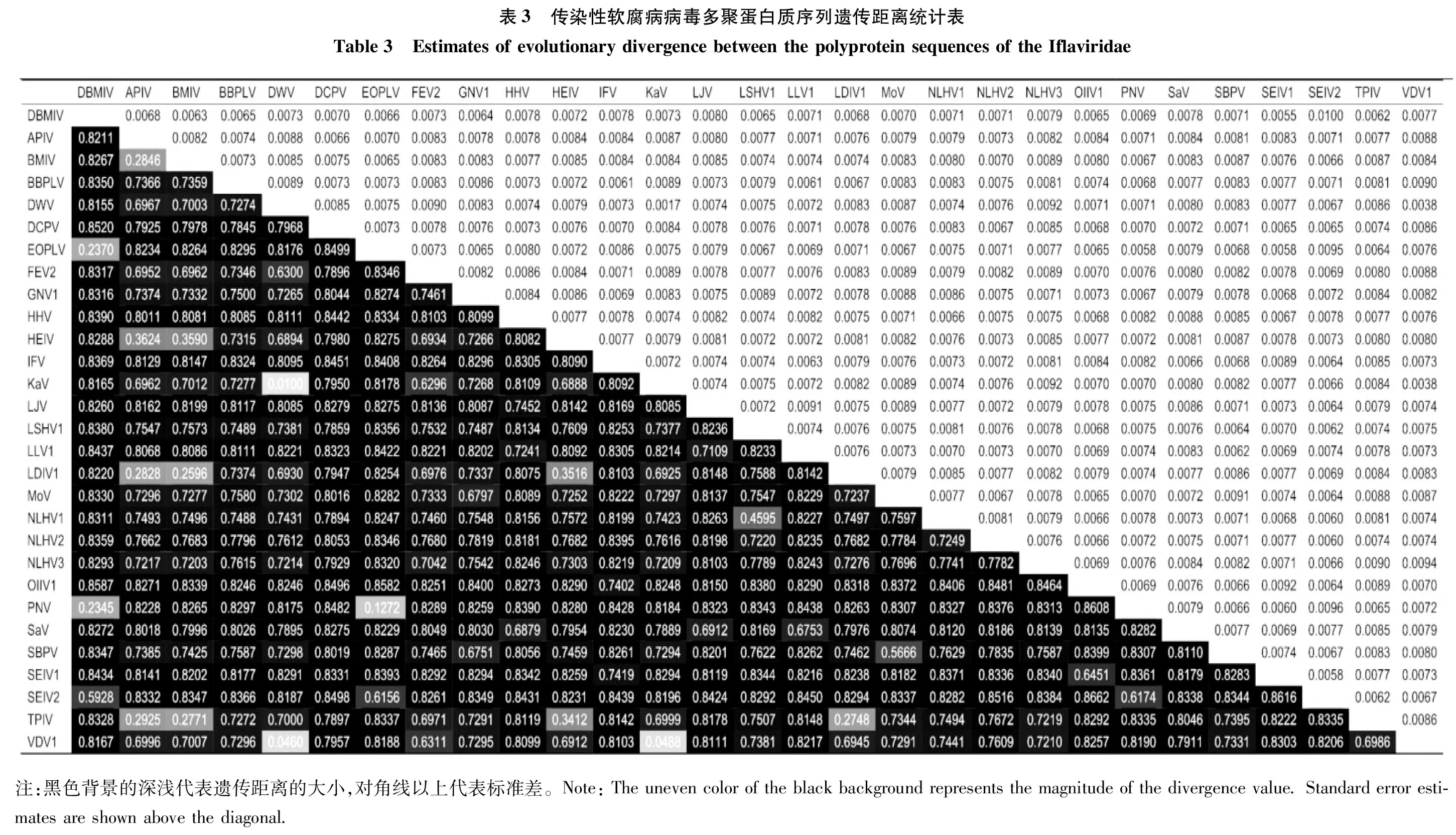
为了确认DBMIV确实为新的病毒,把DBMIV多聚蛋白的氨基酸序列与Iflavirus属的病毒物种的多聚蛋白做比较。多序列比对后,两两比较遗传距离,最终汇总成表3。DBMIV与大多数传染性软腐病病毒遗传距离都较远,但与PNV和EOPLV遗传距离相近,其遗传距离分别为0.2345±0.0069和0.2370±0.0066。PNV和EOPLV之间的遗传距离为0.1272±0.0058。为了确定DBMIV在Iflavirus属的进化关系,利用Iflavirus属的多聚蛋白序列构建的最大似然(ML)树以及贝叶斯推论(BI)树(图5)。两种构建进化树的方法所重构的拓扑结构类似,且大部分的置信值与后验概率很高。但是BBPLV在ML树中形成独立的分支,但置信值很低,在BI树中与DCPV形成姊妹群,后验概率较高(0.95)。DBMIV和PNV、EOPLV、SEIV2形成一个分支且置信值和后验概率都很高。为了确定DBMIV在Iflavirus属的进化地位,利用双顺反子病毒科(Dicistroviridae)作为外群(图6),通过病毒常用的RNA聚合酶基因进行多序列比对后构建ML树和BI树,重构的拓扑结构十分类似,且BBPLV和DCPV形成姊妹群,虽然在ML树中置信值为88,在BI树中后验概率为0.98。通过加入外群后发现DBMIV、PNV、EOPLV和SEIV2形成的分支首先与其他Iflavirus属的病毒分开。
2.4 DBMIV病毒的RT-PCR验证
利用DBMIV基因组序列设计出两对RT-PCR引物DBMIV1和DBMIV2用于扩增DBMIV的CP蛋白区域。对田间采集的小菜蛾样品进行检测后发现两个样品能够获得阳性条带。见图7所示引物DBMIV1和DBMIV2均能获得大于1kb的电泳条带,1号样品条带很亮且测序结果也得到证实,但2号样品的检测条带亮度很弱,也无法得到测序结果,这可能因为DBMIV的滴度在田间某些成虫体内很低有关。另外值得注意的是并非所有样品都扩增出RT-PCR的条带,这说明小菜蛾田间种群的DBMIV感染率并非100%。
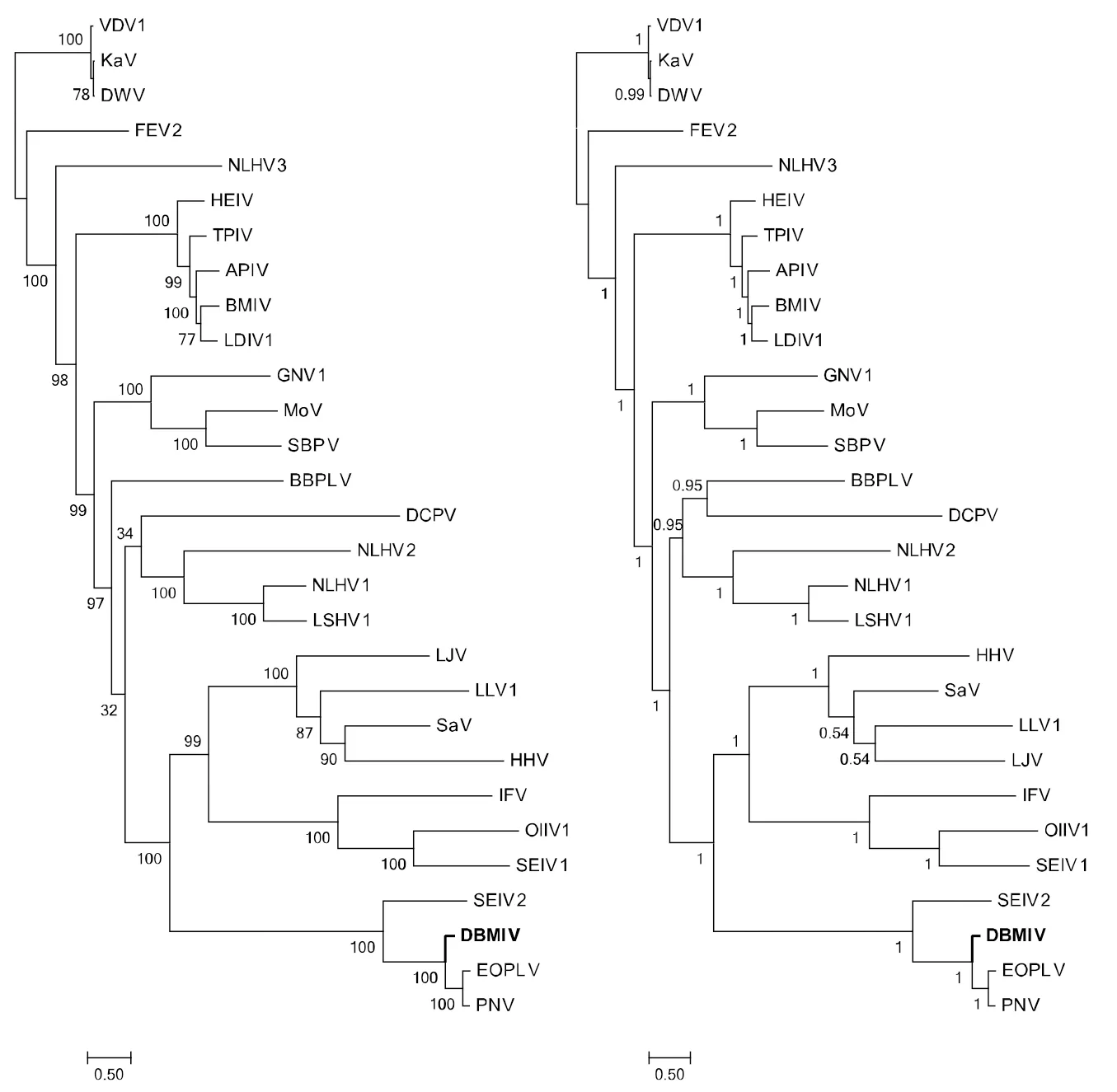
图5 基于多聚蛋白序列构建最大似然法(左)和贝叶斯法(右)进化树Fig.5 Phylogenetic ML (left) and Bayesian (right) tree based on the amino acid sequences of the polyproteins of Iflaviridae
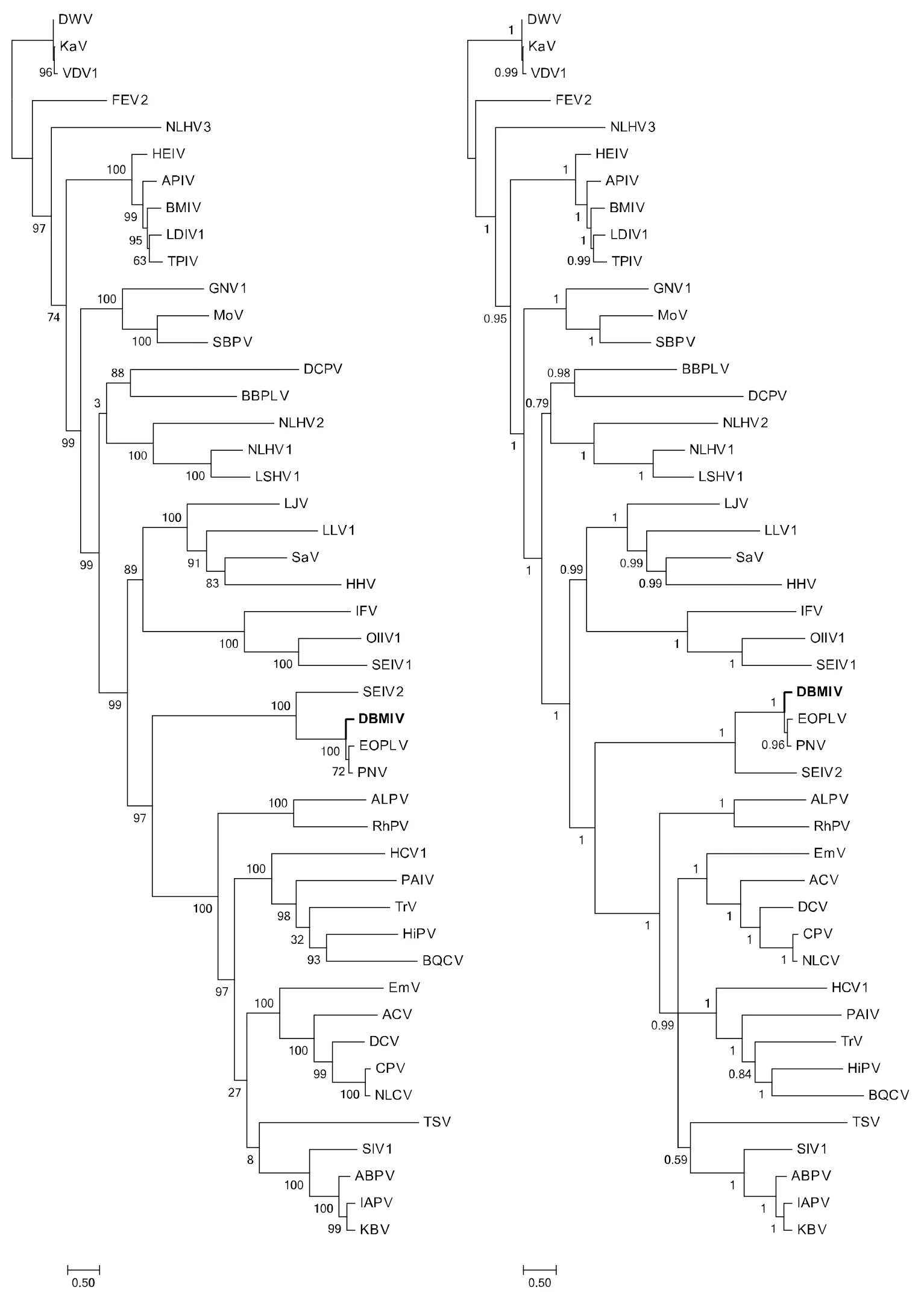
图 6 基于RNA聚合酶蛋白序列构建的最大似然法(左)和贝叶斯法(右)进化树Fig.6 Phylogenetic ML (left) and Bayesian (right) tree based on the amino acid sequences of the RdRp proteins with the outgroup of Dicistroviridae
3 结论与讨论
病毒广泛分布(Dinsdaleetal.,2008;López-Buenoetal.,2009),几乎能感染任何的物种,且在生态系统中扮演重要的角色(Handleyetal.,2012;Ignacio-Espinozaetal.,2013;Xuetal.,2015),甚至在无脊椎动物的转录组序列中也常常能够发现新的病毒物种(Shietal.,2016)。本文报道的是在实验室拼接测序的小菜蛾转录组序列时,获得一条长9580 bp的病毒基因组序列,ORF预测其可编码一条完整的蛋白质且包含病毒衣壳蛋白、RNA酶和解旋酶等序列保守域,其序列特征十分类似传染性软腐病病毒,因此将其命名为小菜蛾传染性软腐病病毒,英文简称DBMIV。随着高通量测序成本的逐年下降以及更多的高通量测序的完成,利用高通量测序将发现并命名更多的病毒种类,如三种褐飞虱蜜露病毒都是利用RNA测序数据拼接而成的(Murakamietal.,2013a;Murakamietal.,2013b)。
核酸的GC含量影响着核酸的稳定性、变异速率以及二级结构等。DBMIV的5′UTR其GC含量比其他区域低,可能因为5′UTR具有IRES位点,需要折叠成一定的二级结构并招募核糖体(Lietal.,2012;Luetal.,2007;Luetal.,2006;Ongusetal.,2006;Wuetal.,2007)。DBMIV的ORF区域并没有严重的核酸偏好,其原因为氨基酸使用偏好没有严重的偏倚,且也没有偏好使用高AT或GC的同义密码子。
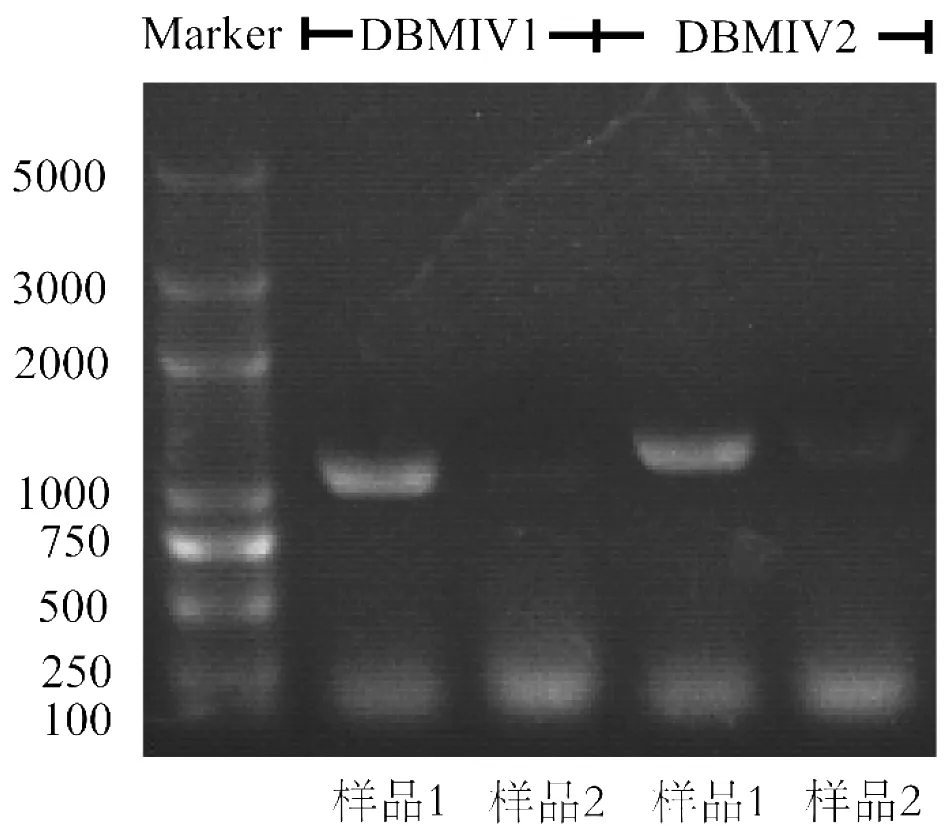
图 7 DBMIV的RT-PCR检测电泳图Fig.7 Electrophoretogram of the RT-PCR detections to DBMIV
将Iflaviridae病毒属的多聚蛋白遗传距离进行计算与比较发现,DBMIV与PNV和EOPLV遗传距离最相近。但是其遗传距离比PNV和EOPLV之间的遗传距离大。DWV和KaV普遍被认为是同种病毒的不同亚型,而VDV1被认为是独立的病毒,DWV和KaV之间的遗传距离为0.0100±0.0017,VDV1与DWV和KaV之间的遗传距离为0.0460±0.0038和0.0488±0.0038,都小于DBMIV与PNV和EOPLV的遗传距离。DBMIV的ORF长9039,与PNV和EOPLV的ORF(8961和8964)相差78和75个氨基酸残基,相差也较大,因此可以确定DBMIV为新病毒种类。
在构建Iflaviridae系统发育关系时,用多聚蛋白全序列与用RNA聚合酶序列构建的进化树的拓扑分支结构十分类似,因此RNA聚合酶序列虽然是病毒基因组的部分序列,但却能够提供足够的进化信息,也因此RNA聚合酶序列常常被用来重构病毒系统发生与进化关系(Ryabov,2007;Shietal.,2016;Wangetal.,2004)。利用多聚蛋白全序列构建的ML树和BI树时,BBPLV的分支结构不一致,BBPLV所在的ML树形成独立分支但分支置信值较低,但是利用多聚蛋白全序列构建的BI树与RNA聚合酶序列构建的进化树分支结构是类似的,因此推测BBPLV与DCPV形成姊妹群,而不是独立的分支。多聚蛋白全序列构建的结果不一致的原因可能为不同病毒的序列长度差别大,保守结构域不连续,产生的空隙(Gaps)较多,因此容易产生进化杂信号而导致的。RNA聚合酶序列较保守,长度变化不大,且是RNA病毒共有的、功能类似的基因,在重构RNA病毒系统发育上具有天然的优势。
许多传染性软腐病病毒能够引起寄主严重的病症,如造成蚕桑业严重经济损失的家蚕软化病毒(IFV)感染家蚕中肠上皮细胞,引起软化腐烂并导致死亡(Ayuzawa,1972)。但也发现一些传染性软腐病病毒种类的感染未发现明显症状。与小菜蛾软腐病病毒遗传进化关系相近的SEIV2未发现对寄主甜菜夜蛾致病(Choietal.,2012),EOPLV被报导从颗粒体病毒感染致死的茶尺蠖幼虫中分离出来的,暗示其与颗粒体病毒协同对茶尺蠖幼虫产生致死作用(Wangetal.,2004)。另一种与小菜蛾软腐病病毒遗传进化关系相近的PNV病毒对寄主榕透翅毒蛾产生类黄疸症状,并且与榕透翅毒蛾核多角体病毒(Perina nuda nucleopolyhedrosis,PenuNPV)协同产生软腐病病症(Wangetal.,1999)。虽然小菜蛾软腐病病毒是利用转录组数据偶然被发现的,但深入研究其对寄主的致病性,或者与其他病毒一起对寄主的协同致病性等生物学问题,可为小菜蛾生物防治提供新思路,对开发小菜蛾病毒性杀虫剂有重要科学意义。
References)
Anisimova M,Gascuel O.Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative [J].SystematicBiology,2006,55(4):539-552.
Asgari S,Johnson KN.Insect virology [M].Australia:Caister Academic Press,2010.
Ayuzawa C.Studies on the infectious flacherie of the silkworm,BombyxmoriL.:I.Purification of the virus and its some properties [J].JournalofInsectBiotechnologyandSericology,1972,41(5):338-344.
Bailey L,Gibbs AJ,Woods RD.Sacbrood virus of the larval honey bee (ApismelliferaLinnaeus) [J].Virology,1964,23:425-429.
Camacho C,Coulouris G,Avagyan V,etal.BLAST+: architecture and applications [J].BMCBioinformatics,2009,10:421.
Choi JY,Kim YS,Wang Y,etal.Complete genome sequence of a novel picorna-like virus isolated fromSpodopteraexigua[J].JournalofAsia-PacificEntomology,2012,15(2):259-263.
Dinsdale EA,Edwards RA,Hall D,etal.Functional metagenomic profiling of nine biomes [J].Nature,2008,452(7187):629-632.
Doudna JA,Sarnow P.Translational Control in Biology and Medicine.Translation initiation by viral internal ribosome entry sites [M].Cold Spring Harbor Monograph Archive,2007,48:129-153.
Fujiyuki T,Takeuchi H,Ono M,etal.Novel insect picorna-like virus identified in the brains of aggressive worker honeybees [J].JournalofVirology,2004,78(3):1093-1100.
Guindon S,Dufayard JF,Lefort V,etal.New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0 [J].SystematicBiology,2010,59(3):307-321.
Haas BJ,Papanicolaou A,Yassour M,etal.Denovotranscript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis [J].NatureProtocols,2013,8(8):1494-1512.
Handley SA,Thackray LB,Zhao G,etal.Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome [J].Cell,2012,151(2):253-266.
Ignacio-Espinoza JC,Solonenko SA,Sullivan MB.The global virome: not as big as we thought? [J].CurrentOpinioninVirology,2013,3(5):566-571.
Kawase S,Hashimoto Y,Nakagaki M.Characterization of flacherie virus of the silkworm,Bombyxmori[J].TheJournalofSericulturalScienceofJapan,1980,49(6):477-484.
Kumar S,Stecher G,Tamura K.MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets [J].MolecularBiologyandEvolution,2016,33(7):1870-1874.
López-Bueno A,Tamames J,Velázquez D,etal.High diversity of the viral community from an antarctic lake [J].Science,2009,326(5954):858-861.
Li M,Man N,Qiu H,etal.Detection of an internal translation activity in the 5′ region ofBombyxmoriinfectious flacherie virus [J].AppliedMicrobiologyandBiotechnology,2012,95(3):697-705.
Lu J,Hu Y,Hu L,etal.Ectropisobliquapicorna-like virus IRES-driven internal initiation of translation in cell systems derived from different origins [J].JournalofGeneralVirology,2007,88(Pt 10):2834-2838.
Lu J,Zhang J,Wang X,etal.Invitroandinvivoidentification of structural and sequence elements in the 5′ untranslated region ofEctropisobliquapicorna-like virus required for internal initiation [J].JournalofGeneralVirology,2006,87(Pt 12):3667-3677.
Murakami R,Suetsugu Y,Kobayashi T,etal.The genome sequence and transmission of an iflavirus from the brown planthopper,Nilaparvatalugens[J].VirusResearch,2013a,176(1-2):179-187.
Murakami R,Suetsugu Y,Nakashima N.Complete genome sequences of two iflaviruses from the brown planthopper,Nilaparvatalugens[J].ArchivesofVirology,2013b,159(3):585-588.
Ongus JR,Peters D,Bonmatin JM,etal.Complete sequence of a picorna-like virus of the genusIflavirusreplicating in the miteVarroadestructor[J].JournalofGeneralVirology,2004,85(Pt 12):3747-3755.
Ongus JR,Roode EC,Pleij CW,etal.The 5′ non-translated region ofVarroadestructorvirus 1 (genusIflavirus): structure prediction and IRES activity inLymantriadisparcells [J].JournalofGeneralVirology,2006,87(Pt 11):3397-3407.
Pestova TV,Kolupaeva VG,Lomakin IB,etal.Molecular mechanisms of translation initiation in eukaryotes [J].ProceedingsoftheNationalAcademyofSciencesoftheUnitedStatesofAmerica,2001,98(13):7029-7036.
Ronquist F,Teslenko M,van der Mark P,etal.MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space [J].SystematicBiology,2012,61(3):539-542.
Ryabov EV.A novel virus isolated from the aphidBrevicorynebrassicaewith similarity to Hymenoptera picorna-like viruses [J].JournalofGeneralVirology,2007,88(Pt 9):2590-2595.
Shi M,Lin XD,Tian JH,etal.Redefining the invertebrate RNA virosphere [J].Nature,2016,540(7634):539-543.
Simmonds P,Adams MJ,Benko M,etal.Consensus statement: Virus taxonomy in the age of metagenomics [J].NatureReviewsMicrobiology,2017,15(3):161-168.
Stöver BC,Müller KF.TreeGraph 2: combining and visualizing evidence from different phylogenetic analyses [J].BMCBioinformatics,2010,11:7.
van Oers MM.Insect virology.Genomics and biology of Iflaviruses [M].Australia:Caister Academic Press,2010,10:231-250.
Wang CH,Wu CY,Lo CF.A new picorna-like virus, PnPV, isolated from ficus transparent wing moth,Perinanuda(Fabricius) [J].JournalofInvertebratePathology,1999,74(1):62-68.
Wang X,Zhang J,Lu J,etal.Sequence analysis and genomic organization of a new insect picorna-like virus,Ectropisobliquapicorna-like virus, isolated fromEctropisobliqua[J].JournalofGeneralVirology,2004,85(Pt 5):1145-1151.
Wu CY,Lo CF,Huang CJ,etal.The complete genome sequence ofPerinanudapicorna-like virus, an insect-infecting RNA virus with a genome organization similar to that of the mammalian picornaviruses [J].Virology,2002,294(2):312-323.
Wu TY,Wu CY,Chen YJ,etal.The 5′ untranslated region ofPerinanudavirus (PnV) possesses a strong internal translation activity in baculovirus-infected insect cells [J].FebsLetters,2007,581(16):3120- 3126.
Xu GJ,Kula T,Xu Q,etal.Comprehensive serological profiling of human populations using a synthetic human virome [J].Science,2015,348(6239):aaa0698.
Yue C,Genersch E.RT-PCR analysis of Deformed wing virus in honeybees (Apismellifera) and mites (Varroadestructor) [J].JournalofGeneralVirology,2005,86(Pt 12):3419-3424.
Phylogenetics analysis of the complete genome of Diamondback moth iflavirus
CHEN Da-Song, DAI Jian-Qing*, HAN Shi-Chou
(Guangdong Key Laboratory of Animal Conservation and Resource Utilization, Guangdong Public Laboratory of Wild Animal Conservation and Utilization, Guangdong Institute of Applied Biological Resources, Guangzhou 510260, China)
The growing number of Iflaviruses are positive strind RNA viruses, which belong to Picornavirales.The hosts of Iflaviruses include Lepidoptera, Hymenoptera, Hemiptera andVarroadestructor.Iflaviruses are pathogenic to some main agricultural pests and are the potential viruses that could be used in biological control.In this study we assembled a complete Iflavirus genome from the transcriptome data of diamond moth.We find an ORF in this genome which can encode a complete polyprotein homologue to other Iflavirus genomes.Therefore, we nominate this new virus as Diamondback moth iflavirus, and the abbreviation is DBMIV.The phylogenetic analysis based on the polyprotein and RNA polymerase sequences showed that the DBMIV was evolutionally closed to PNV, EOPLV and SEIV2.Eventually, DBMIV was verified to infect the DBM in the field by RT-PCR amplification of the coat protein region.The identification of the DBMIV provided a referential scheme of virus species identification from high-through sequencing data, and offered a new research approach of virus control to the diamond moth.
Plutellaxylostella; Iflavirus; genome; phylogenetics
陈大嵩,戴建青,韩诗畴.小菜蛾传染性软腐病病毒的全基因组序列及其进化分析[J].环境昆虫学报,2017,39(3):618-631.
国家自然科学基金(31371932);广东省科学院青年科学研究基金(qnjj201602);广东省科学院科技发展专项(2017GDASCX-0107)
陈大嵩,男,1986年生,博士,主要从事分子生物学和化学生态学研究,E-mail: chends@giabr.gd.cn
*通讯作者Author for correspondence, E-mail: jqdai@giabr.gd.cn
Received: 2017-02-10;接受日期Accepted: 2017-05-18
Q968.1;S433.4
A
1674-0858(2017)03-0618-14
猜你喜欢
植物保护(2022年1期)2022-02-10
农业技术与装备(2022年11期)2022-01-01
上海农业科技(2020年5期)2020-10-24
生物学通报(2020年11期)2020-10-22
发明与创新·中学生(2019年6期)2019-06-26
蔬菜(2018年9期)2018-09-21
中成药(2018年7期)2018-08-04
现代园艺(2017年13期)2018-01-19
农民致富之友(2017年7期)2017-04-27
湖北农业科学(2016年23期)2017-03-17
