
DNA条形码专用R包及其主要功能简介
2017-08-02 01:39阮倩倩罗桂杰张爱兵
环境昆虫学报 2017年3期
金 倩,阮倩倩,陈 芬,罗桂杰,张爱兵
DNA条形码专用R包及其主要功能简介
金 倩1,2,阮倩倩1,陈 芬1,罗桂杰1,张爱兵2*
(1.江苏省农业科学院宿迁农科所,江苏宿迁 223800;2.首都师范大学生命科学学院,北京 100048)
SPIDER和BarcodingR软件包作为DNA条形码研究的专用R软件包,整合了基于DNA条形码的物种识别算法和数据分析策略,避免了各种非商业用途软件的局限性,为DNA条形码的研究提供了便捷性和可操作性。本文对SPIDER和BarcodingR软件包的主要功能进行整体介绍,并以西藏夜蛾科COI、松毛虫属ITS等数据为例,进行主要函数的演示分析,希望为昆虫DNA条码的应用分析提供帮助。
DNA条形码;R语言;SPIDER;BarcodingR
近年来,伴随着DNA条形码的广泛应用和后基因组时代的到来,生物多样性的研究变得更加深入和便捷。截至2016年12月30日,BOLD系统中共记录物种261170个,条形码序列5281492条(http://www.barcodinglife.org)。在DNA条形码数据快速增长的背景下,物种识别和构建类群全面的参考数据库成为生物多样性研究的核心任务,进而推动了物种识别算法等理论研究的不断发展。基于DNA条形码的识别方法及有效性,包括经典的基于距离、基于进化树和基于特征的方法(Sarkaretal., 2002;Blaxteretal., 2005;Toffolietal., 2008)。近年来研究者也试图提出各种新的方法,如,人工智能方法(Zhangetal., 2012a)、模糊集合理论(Zhangetal., 2012b)、构建识别效率与距离阈值之间模型的方法(Virgilioetal., 2012)、基于贝叶斯理论和形态有限介入的方法(Jinetal., 2013)等。
上述理论方法的实现对用户的生物信息学背景要求较高,有些软件因缺乏友好的用户界面或涉及非商业用途而被限制使用,对试图通过DNA条形码技术研究生物类群的实例研究者而言,复杂的算法和程序往往使他们望而生畏。SPIDER软件包(Brownetal., 2012)和BarcodingR软件包(Zhangetal., 2016)的相继提出避免了以上非商业用途软件的局限性,发挥R语言本身在数据处理、数据统计和数据可视化方面较强的优势,为DNA条形码的研究提供了便捷性和可操作性。本文对SPIDER和BarcodingR软件包的主要功能进行系统的介绍,并以西藏夜蛾科COI(Jinetal., 2013)、松毛虫属ITS(Daietal., 2013)等数据为例,进行主要函数的演示分析。
1 SPIDER软件包的主要函数及功能
SPIDER软件包以软件包“APE”(Paradisetal., 2004),“PHYCLUST”(Chen and Dorman, 2010),“PEGAS”(Paradis, 2010)和“ADEGENET”(Jombart, 2008)为基础建立,涉及到条形码分析的主要函数及功能详见表1。该软件包首先可以对序列的属性进行统计,例如可以根据dataStat函数计算数据的属、种及样本数量;其次,能够实现经典的基于距离树的物种识别效率计算,需要注意的是数据集中的所有序列必须事先进行识别鉴定,并建立参考条形码数据库。在计算识别效率时,每一条序列被作为未知序列问询数据库中参考序列,如果识别的结果与已知结果一致,表明识别成功,否则为失败;最后,该软件包还可以检验系统发育树的单系性,能够在Genbank和BOLD数据库中直接获取数据,实现滑动窗口分析等功能。

表1 SPIDER软件包主要函数及功能Table 1 List of main functions and their descriptions of the ‘SPIDER’ package
2 BarcodingR软件包的主要函数及功能
BarcodingR软件包以软件包“APE”(Paradisetal., 2004),“ADEGENET”(Jombart, 2008),“MCLUST”(Fraley and Raftery, 1999),“PHYCLUST”(Chen and Dorman, 2010),“CCLUST” (https://cran.r-project.org/),“PEGAS”(Paradis, 2010),“SEQINR”(Charifetal., 2005),“NNET” (Ripley, 1996),“E1071”(https://cran.r-project.org/)和“SPIDER”(Brownetal., 2012)为基础建立。该软件包主要用途是进行物种识别和条形码分析。具体函数及功能详见表2,其中3个较为关键的物种识别函数依次是‘barcoding.spe.identify’,‘barcoding.spe.identify2’ 和‘bbsik’。
barcoding.spe.identify函数进行物种识别主要通过人工智能方法、模糊集合理论和贝叶斯方法,通过以下参数进行控制:
2.1 人工智能方法bpNewTraining/bpNewTrainingOnly/bpUseTrained参数
当数据集较小时(<500条序列),可以选择bpNewTraining参数,训练集和问询集同时计算。当数据集较大时(>500条序列)需要通过bpNewTrainingOnly参数先进行人工智能训练,然后再通过bpUseTrained进行问询集的物种识别。训练的模型参数被存放于一个临时文件中,当运行bpUseTrained时加载训练结果并进行物种识别。
2.2 模糊集合理论fuzzyId参数
每一条问询序列的识别结果都被赋予一个FMF值(范围从0~1),代表序列分配的可能性(Zhangetal., 2012a)。为了提高识别效率,在搜索参考数据过程中采用KNN搜索算法替代MD算法(http://www.stats.ox.ac.uk/pub/MASS4/)。
2.3 贝叶斯方法Bayesian参数
按照金倩等(Jinetal., 2013)提出的基于贝叶斯理论和形态有限介入的方法对蛾类进行物种识别,主要通过Bayesian函数实现。

表2 BarcodingR软件包主要函数及功能Table 2 List of main functions and their descriptions of the ‘BarcodingR’ package
除了COI序列之外,非编码基因ITS1和ITS2也被广泛应用于动植物类群中。但因其间隔区的存在影响了多序列比对的准确性(Zhangetal., 2008;Zhangetal., 2012b),故张爱兵等(Zhangetal., 2012b)提出在非编码基因中可以免除序列比对过程直接进行物种识别,但该方法受限于MATLAB环境下使用。BarcodingR软件包规避了语言环境的局限性,通过kmer方法实现非编码基因的序列比对和识别。
3 应用实例分析
3.1 数据输入
SPIDER软件包可以通过ape软件包中read.dna函数read.nexus.data函数读入分子数据,通过R基础包中的read.table读入形态数据,也可以加载软件包本身的数据。以读入软件包中数据和用户自己的数据为例,R语言的命令代码如下,#后面的文字表示对R语言命令的注释。
>library(spider) #加载spider软件包
>data(dolomedes) #加载dolomedes数据
>setwd("c:\My documents\R") #设定数据集所在路径
>dataset <- read.dna("mySequences.fas",format="fasta") #读入"mySequences.fas"数据
BarcodingR软件包的输入较复杂,需提前准备好参考序列和问询序列,格式为常用的.fasta或.fas格式文件。参考序列的.fasta文件大于号之后必须含有分类信息“>seqID, species_names”,例如:“>seq1, Noctuidae_Blepharosis_paspa”,“>seq2,Apamea_devastator”。问询序列为一般的.fasta格式,例如“>seqID”。通过ape软件包中read.dna函数或adegent软件包中fasta2DNAbin函数读入数据(Paradisetal., 2004)。
>library(ape) #加载ape软件包
>ref.dna <- read.dna("ref.fas",format="fasta") #读入参考序列
>library(adegenet) #加载adegenet软件包
>que.dna <- fasta2DNAbin("que.fas") #读入问询序列
3.2 从GenBank和BOLD数据库中下载数据
APE软件包中的read.GenBank函数能够直接下载GenBank中的序列,并存储为DNAbin格式的对象中,但该函数不能获得物种名字或者基因区域等信息,因此SPIDER软件包中的read.GB函数对其进行了改进,以GenBank的登录号为向量,并检索记录对应的序列。以西藏夜蛾数据为例(Jinetal., 2013),在GenBank中下载序列步骤如下:
>seq <- 392408:392727 #将392408至392727编号赋值给seq
>seq <- paste("JX", seq, sep="") #在每个编号前添加"JX"
>TibetData <- read.GB(seq) #下载数据并赋值给TibetData
SPIDER软件包中的search.BOLD函数在BOLD数据库中搜寻某类群的条形码数据,并返回搜寻对象的样本编号,read.BOLD函数通过这些样本编号下载对应的序列。以查找夜蛾科物种Himalaeaunica为例(Jinetal., 2013),R语言的命令代码如下:
>nums <- search.BOLD("Himalaeaunica") #在BOLD数据库中查找夜蛾科Himalaea
#unica类群并将样本编号返回给nums
>NoctuidaeSp <- read.BOLD(nums) #下载mums对应的序列并赋值给NoctuidaeSp
3.3 基于距离的物种识别效率分析
以SPIDER软件包中Anoteropsis数据为例(Vink and Paterson, 2003),进行threshID函数计算,R语言的命令如下,结果如图1所示,“correct”代表在1%的阈值范围内问询序列识别正确的是11个,“incorrect”代表在1%的阈值范围内问询序列识别错误的是2个,“no id”代表在1%的阈值范围内问询序列没有匹配到任何一个个体是20个。
>library(spider) #加载spider软件包
>data(anoteropsis) #加载anoteropsis数据
>aa <- strsplit(dimnames(anoteropsis)[[1]], split="_") #将序列名字按照“_”分开
>anoSpp <- sapply(aa, function(x) paste(x[1], x[2], sep="_")) #将属名种名提取出来
>inputDist<-dist.dna(anoteropsis,pairwise.deletion=TRUE) #计算K2P距离
>table(threshID(inputDist,anoSpp)) #1%阈值范围内进行物种识别效率计算
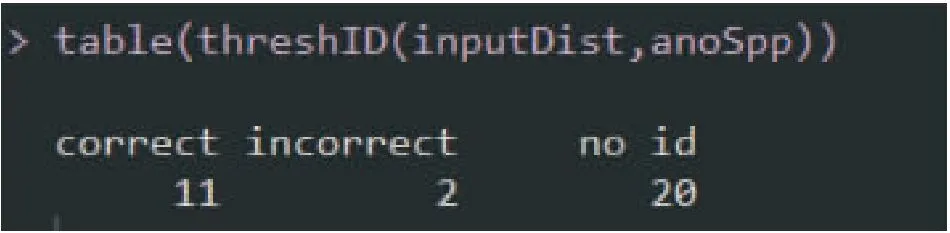
图1 基于1%阈值的物种识别结果Fig.1 Species identification results with 1% threshold
3.4 物种识别分析
以BarcodingR软件包中的夜蛾科数据为例(Jinetal., 2013)。首先需要加载软件包和数据,然后通过控制barcoding.spe.identify函数中的method参数控制不同的物种识别算法,R语言的命令如下:
>install.packages("BarcodingR") #安装BarcodingR软件包
>library(BarcodingR) #加载BarcodingR软件包
>refdata<- TibetanMoth #获取 TibetanMoth数据集并赋值给变量refdata
>output<- sample.ref(refdata, sample.porp = 0.9,sample.level = "species") #随机抽样
>ref<- output$ref.selected #定义参考数据集
>que<- out$ref.left #定义问询数据集
>bp<- barcoding.spe.identify(ref,que,method ="bpNewTraining") #人工智能物种识别
>save.ids(outfile="BPidentified.txt",bp) #输出结果
#输出结果BPidentified.txt详见附件1
>fuzzyID<- barcoding.spe.identify(ref,que,method ="fuzzyId") #模糊集合物种识别
>save.ids(outfile="fuzzyIDidentified.txt",fuzzyID) #输出结果
#输出结果fuzzyIDidentified.txt详见附件2
>Bay<- barcoding.spe.identify(ref,que,method ="Bayesian") #贝叶斯物种识别
>save.ids(outfile="Bayidentified.txt",Bay) #输出结果
#输出结果Bayidentified.txt详见附件3
3.5 不同物种识别方法结果的一致性检验及汇总
通过BarcodingR软件包中的函数consensus.identify,实现人工智能方法、模糊集合理论和贝叶斯方法的一致性检验,以西藏夜蛾数据为例(Jinetal., 2013),不同方法的物种一致性检验R命令如下,结果详见表3。
>ref<-TibetanMoth #获取 TibetanMoth数据集并赋值给变量ref
>set.seed(10) #产生随机数
>out<-sample.ref(ref,sample.porp=0.95,sample.level="species") #随机抽样
>ref2<-out$ref.selected #定义参考数据集
>que<-out$ref.left #定义问询数据集
>bsi0<-barcoding.spe.identify(ref2, que, method ="bpNewTrainingOnly") #人工智能物种
#识别
>bsi1<-barcoding.spe.identify(ref2, que, method ="bpUseTrained") #人工智能物种识别
>bsi2<-barcoding.spe.identify(ref2, que, method ="fuzzyId") #模糊集合物种识别
>bsi3<-barcoding.spe.identify(ref2, que, method ="Bayesian") #贝叶斯物种识别
>que.IDs<-gsub(",.+","",rownames(que)) #提取序列ID
>bpid<-bsi1$output_identified$spe.Identified #将人工智能物种识别结果赋值
>fuzzyid<-bsi2$output_identified$spe.Identified #将模糊集合物种识别结果赋值
>Bayesianid<-bsi3$output_identified$output_identified #将贝叶斯物种识别结果赋值
>identifications<-data.frame(queIDs = que.IDs, pid = bpid, fuzzyid = fuzzyid, Bayesianid = Bayesianid) #将三种方法的物种识别结果输出放入数据框identifications中
>ccs<-consensus.identify(identifications) #一致性检验

表3 基于不同的物种识别方法的物种一致性Table 3 List of main functions and their descriptions of the ‘BarcodingR’ package
续上表
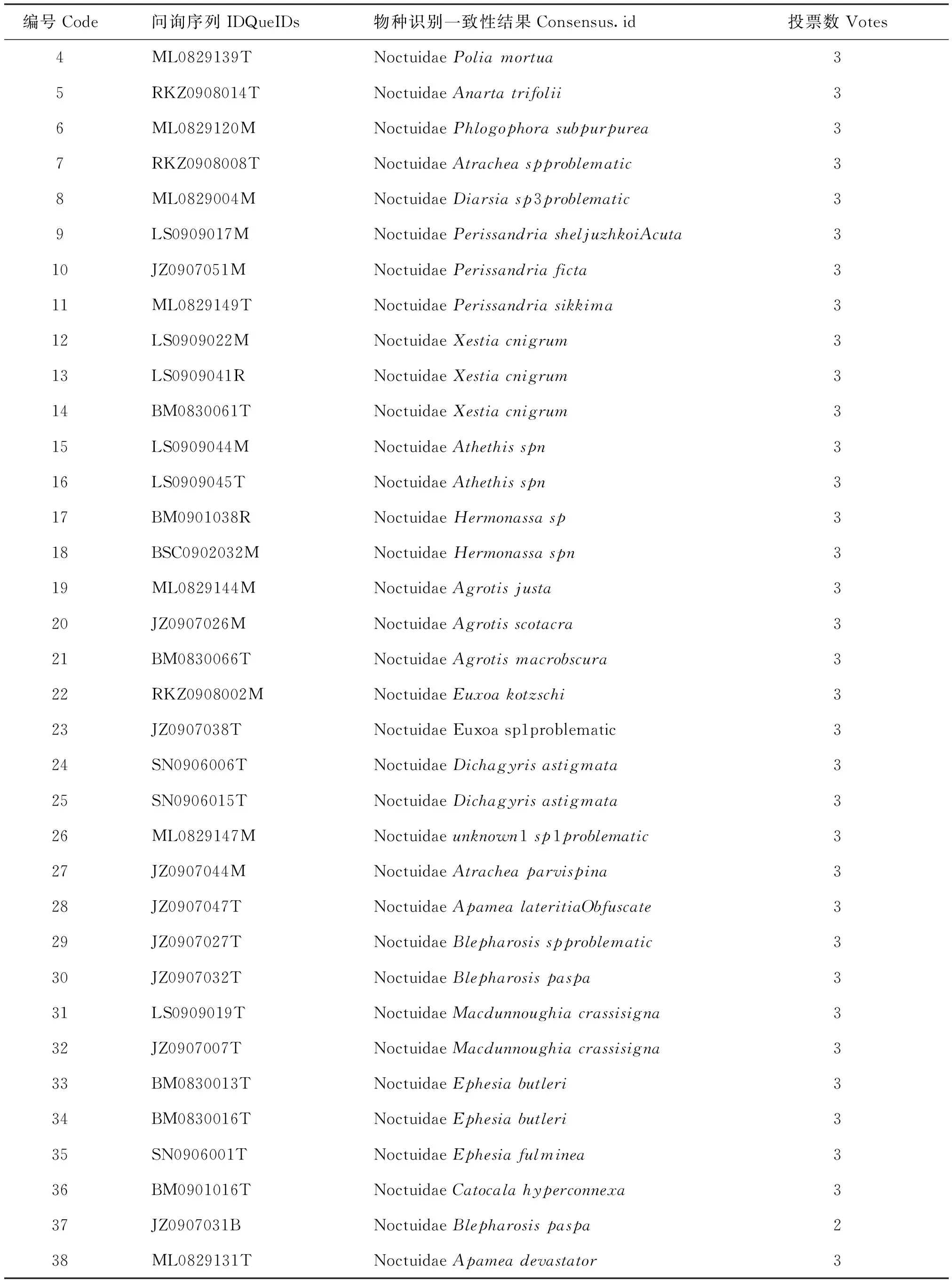
编号Code问询序列IDQueIDs物种识别一致性结果Consensus.id投票数Votes4ML0829139TNoctuidaePoliamortua35RKZ0908014TNoctuidaeAnartatrifolii36ML0829120MNoctuidaePhlogophorasubpurpurea37RKZ0908008TNoctuidaeAtracheaspproblematic38ML0829004MNoctuidaeDiarsiasp3problematic39LS0909017MNoctuidaePerissandriasheljuzhkoiAcuta310JZ0907051MNoctuidaePerissandriaficta311ML0829149TNoctuidaePerissandriasikkima312LS0909022MNoctuidaeXestiacnigrum313LS0909041RNoctuidaeXestiacnigrum314BM0830061TNoctuidaeXestiacnigrum315LS0909044MNoctuidaeAthethisspn316LS0909045TNoctuidaeAthethisspn317BM0901038RNoctuidaeHermonassasp318BSC0902032MNoctuidaeHermonassaspn319ML0829144MNoctuidaeAgrotisjusta320JZ0907026MNoctuidaeAgrotisscotacra321BM0830066TNoctuidaeAgrotismacrobscura322RKZ0908002MNoctuidaeEuxoakotzschi323JZ0907038TNoctuidaeEuxoasp1problematic324SN0906006TNoctuidaeDichagyrisastigmata325SN0906015TNoctuidaeDichagyrisastigmata326ML0829147MNoctuidaeunknown1sp1problematic327JZ0907044MNoctuidaeAtracheaparvispina328JZ0907047TNoctuidaeApamealateritiaObfuscate329JZ0907027TNoctuidaeBlepharosisspproblematic330JZ0907032TNoctuidaeBlepharosispaspa331LS0909019TNoctuidaeMacdunnoughiacrassisigna332JZ0907007TNoctuidaeMacdunnoughiacrassisigna333BM0830013TNoctuidaeEphesiabutleri334BM0830016TNoctuidaeEphesiabutleri335SN0906001TNoctuidaeEphesiafulminea336BM0901016TNoctuidaeCatocalahyperconnexa337JZ0907031BNoctuidaeBlepharosispaspa238ML0829131TNoctuidaeApameadevastator3
3.6 非编码基因的物种识别
以BarcodingR软件包中的非编码基因数据ITS1为例(Daietal., 2013),通过函数barcoding.spe.identify2实现,R语言的命令如下:
>refdata<- pineMothITS1 #获取pineMothITS1数据集并赋值给变量refdata
>out<- sample.ref(refdata,sample.porp = 0.9,sample.level="species") #随机抽样
>ref<- out$ref.selected #定义参考数据集
>que<- out$ref.left #定义问询数据集
>ITS1identified<- barcoding.spe.identify2(ref,que,kmer=10,optimization="T") #基于kmer
#的模糊集合理论物种识别
>save.ids(outfile="ITS1identified.txt",ITS1identified) #输出结果
#输出结果ITS1identified.txt详见附件4
3.7 DNA条形码空白区
通过函数barcoding.gap实现,该函数输出结果为DNA条形码空白区(如图2),同时还能显示种内和种间距离的最大值、最小值、中位数和平均值。R语言的命令如下:
>b.gap <- barcoding.gap(ref = TibetanMoth, dist = “K80”) #计算种内种间距离并绘制条
#形码空白区图
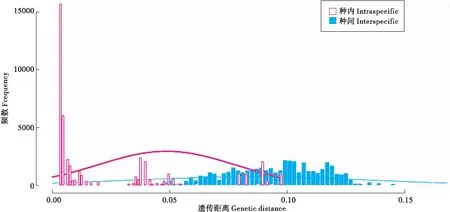
图2 DNA条形码空白区Fig.2 DNA barcoding gap
4 结论
SPIDER软件包(Brownetal., 2012)可以进行常规的条形码统计分析,能够实现经典的基于距离的物种识别效率计算。BarcodingR软件包(Zhangetal., 2016)是SPIDER的后续补充,将近年来新提出的物种识别方法进行了整合,包括人工智能方法、模糊集合理论、基于贝叶斯理论和形态有限介入的方法。两个软件包开发利用R语言平台的免费性、开源性和可扩展性为DNA条形码的研究提供了便捷。SPIDER软件包和BarcodingR软件包都可以从网站中直接下载并使用,SPIDER软件包的网址为http://spider.r-forge.r-project.org/,BarcodingR软件包的网址为:https://cran.r-project.org/web/packages/BarcodingR/index.html或者https://github.com/zhangab2008/BarcodingR.git。二者都适用于Microsoft Windows,MAC OSX 和LINUX系统。
References)
Brown SDJ, Collins RA, Boyer S,etal.SPIDER: An R package for the analysis of species identity and evolution, with particular reference to DNA barcoding [J].MolecularEcologyResources, 2012, 12 (3): 562-565.
Charif D, Thioulouse J, Lobry JR,etal.Online synonymous codon usage analyses with the ade4 and seqinR packages [J].Bioinformatics, 2005, 21 (4): 545-547.
Dai QY, Gao Q, Wu CS,etal.Phylogenetic reconstruction and DNA barcoding for closely related pine moth species (Dendrolimus) in China with multiple gene markers [J].PLoSONE, 2012, 7 (4): e32544.
Fraley C, Raftery AE.MCLUST: Software for model-based cluster analysis [J].JournalofClassification, 1999, 16 (2): 297-306.
Jin Q, Han HL, Hu XM,etal.Quantifying species diversity with a DNA barcoding-based method: Tibetan moth species (Noctuidae) on the Qinghai-Tibetan Plateau [J].PLoSONE, 2013, 8 (5): e64428.
Jombart T.adegenet: A R package for the multivariate analysis of genetic markers [J].Bioinformatics, 2008, 24 (11): 1403-1405.
Paradis E, Claude J, Strimmer K.APE: Analyses of phylogenetics and evolution in R language [J].Bioinformatics, 2014, 20 (2): 289-290.
Paradis E.pegas: An R package for population genetics with an integrated-modular approach [J].Bioinformatics, 2010, 26 (3): 419-420.
Ripley BD.Pattern Recognition and Neural Networks [M].New York: Cambridge University Press, 1996.
Vink CJ, Paterson AM.Combined molecular and morphological phylogenetic analyses of the New Zealand wolf spider genusAnoteropsis(Araneae: Lycosidae) [J].MolecularPhylogeneticsandEvolution, 2003, 28 (3): 576-587.
Zhang AB, Feng J, Ward RD,etal.A new method for species identification via protein-coding and noncoding DNA barcodes by combining machine learning with bioinformatics methods [J].PLoSONE, 2012b, 7 (2): e30986.
Zhang AB, Hao MD, Yang CQ,etal.BarcodingR: An integrated R package for species identification using DNA barcodes [J].MethodsinEcologyandEvolution, 2016: 1-8.
Zhang AB, Muster C, Liang HB,etal.A fuzzy-set-theory-based approach to analyse species membership in DNA barcoding [J].MolecularEcology, 2012a, 21 (8): 1848-1863.
Zhang AB, Sikes DS, Muster C,etal.Inferring species membership using DNA sequences with back-propagation neural networks [J].SystematicBiology, 2008, 57 (2): 202-215.
Main functions and descriptions of R packages used for DNA barcoding
JIN Qian1,2, RUAN Qian-Qian1, CHEN Fen1, LUO Gui-Jie1, ZHANG Ai-Bing2*
(1.Suqian Institute of Agricultural Sciences, Jiangsu Academy of Agricultural Sciences, Suqian 223800, Jiangsu Province, China; 2.College of Life Sciences, Capital Normal University, Beijing 100048, China)
SPIDERand BarcodingR, are new R packages implementing a number of useful functions for DNA barcoding analyses and associated research into species speciation.Both of them avoid limitations of noncommercial computer programs and provide convenience and operability for user.Here we demonstrated the main functions of the two packages with Tibet moth, pine moth datasets.
DNA barcoding; R language; SPIDER; BarcodingR

特邀稿件InvitedReview
金倩,阮倩倩,陈芬,等.DNA条形码专用R包及其主要功能简介[J].环境昆虫学报,2017,39(3):485-492.
国家自然科学基金青年科学基金项目(31601877)
金倩,女,1987年生,博士研究生,研究方向为物种界定及昆虫多样性,E-mail: jinhongyu2001@163.com
*通讯作者Author for correspondence,E-mail: zhangab2008@mail.cnu.edu.cn
Received: 2017-03-01;接受日期Accepted: 2017-04-27
Q963
A
1674-0858(2017)03-0485-08
猜你喜欢
少年文艺·开心阅读作文(2021年8期)2021-09-05
法律方法(2021年4期)2021-03-16
小学科学(学生版)(2019年5期)2019-05-21
少儿美术(快乐历史地理)(2019年11期)2019-04-20
计算机与生活(2018年8期)2018-08-15
小学生导刊(2017年13期)2017-06-15
中学生数理化·高一版(2017年1期)2017-04-25
铁道通信信号(2016年6期)2016-06-01
理科考试研究·高中(2016年9期)2016-05-14
北京信息科技大学学报(自然科学版)(2016年6期)2016-02-27
