
固相萃取/超高效液相色谱-串联质谱法快速测定不同水体中5种人造甜味剂、6种造影剂及咖啡因
2017-08-01 12:46:33杨愿愿应光国赵建亮刘有胜柳王荣
分析测试学报 2017年7期
杨愿愿,应光国,赵建亮,刘有胜,柳王荣
(1.中国科学院 广州地球化学研究所 有机地球化学国家重点实验室,广东 广州 510640; 2.中国科学院大学,北京 100049)
固相萃取/超高效液相色谱-串联质谱法快速测定不同水体中5种人造甜味剂、6种造影剂及咖啡因
杨愿愿1,2,应光国1*,赵建亮1,刘有胜1,柳王荣1
(1.中国科学院 广州地球化学研究所 有机地球化学国家重点实验室,广东 广州 510640; 2.中国科学院大学,北京 100049)
建立了快速检测不同水样(地表水、污水处理厂进水和出水)中5种人造甜味剂、6种造影剂和咖啡因的超高效液相色谱-串联质谱(UPLC-MS/MS)方法。利用HR-X固相萃取柱对过滤后的水样进行富集净化。采用Zorbax SB-C18色谱柱分离,电喷雾正模式(ESI+)和负模式(ESI-)下分别以乙腈-水(10 mmol/L乙酸铵,0.01%甲酸)和乙腈-水(5 mmol/L乙酸铵,1 mmol/L Tris)为流动相,多反应监测模式(MRM)检测。各目标化合物在5~5 000 μg/L范围内线性关系良好,相关系数均大于0.998。地表水、污水处理厂进水和出水的回收率为70%~120%,相对标准偏差(RSD)为0.8%~19.0%,方法检出限(MDL)分别为0.43~13.8、1.80~34.8、2.30~134 ng/L。应用该方法对广州某污水处理厂进水和出水及受纳河流河水进行检测,除阿斯巴甜、碘他拉酸、碘普罗胺、碘美普尔外,其他物质均有检出且浓度较高。该方法准确、灵敏、操作简便,适用于多种环境水样中人造甜味剂、造影剂和咖啡因的分析检测。
人造甜味剂;造影剂;咖啡因;超高效液相色谱-串联质谱;水样
污水处理厂出水中存在的多种有机污染物对水生生物及人体健康存在潜在危害,因此,污水排放对水环境的污染及其影响受到人们越来越多的关注。污水中有些化合物可以指示或示踪污水排放,因此污水示踪剂是研究污水污染程度的有效工具。目前,已有研究表明人造甜味剂[1-2]、造影剂[3]和咖啡因[4-5]是较为理想的污水示踪剂。
人造甜味剂作为一种食品添加剂,多应用于食品和饮料中,因此针对其在食品和饮料中的检测方法已有大量报道[6-9]。近年来,人造甜味剂作为污水示踪剂引起了人们的广泛关注,针对其在环境介质中的检测方法也相继被开发[10-11];造影剂是一类诊断用药,可用于血管、内脏等体内软组织的医学成像,目前已建立了多种环境中造影剂的检测方法[12-13];咖啡因作为一种兴奋剂多存在于茶叶、咖啡、功能饮料、食品和药品中,其主要检测方法有液相色谱法(HPLC)[14]、气相色谱-质谱联用法(GC-MS)[15]及液相色谱-串联质谱法(LC-MS/MS)[16]。总体而言,上述3类化合物的使用范围和理化性质差异较大,且其水溶性较大,提取、分析难度较大。因此其在环境介质中的同时检测方法鲜有报道。然而在筛选污水示踪剂时,需对这3类物质在环境中的分布进行全面调查。为简化操作、缩短分析时间以及节省分析成本,需要开发一种能够同时检测不同水体中人造甜味剂、造影剂和咖啡因的分析方法。
本研究利用固相萃取对水样进行前处理,采用UPLC-MS/MS对样品进行检测,建立了能够同时检测不同水样中人造甜味剂、造影剂和咖啡因的方法,以期为污水示踪剂的筛选提供准确高效的检测手段。
1 实验部分
1.1 仪器、试剂与材料
Agilent 1200 UPLC/6460 Triple Quard MS超高效液相色谱-串联四极杆质谱联用仪(美国Agilent公司);固相萃取装置(美国Alltech公司);MTN-2800D氮吹仪(天津奥特赛恩斯仪器有限公司)。
标准品:安赛蜜(ACE)、甜蜜素(CYC)、阿斯巴甜(ASP)、三氯蔗糖(SUC)、泛影酸(DTZ)、碘帕醇(IOD)、碘普罗胺(IOP)、碘美普尔(IOM)、碘海醇(IOX)、咖啡因(CAF)购于德国Dr. Ehrenstorfer公司,糖精钠(SAC)购于美国Supelco公司,碘他拉酸(ITA)购于美国药典委员会(USP);氘代内标:安赛蜜-d4(ACE-d4)、阿斯巴甜-d5(ASP-d5)、糖精-d5(SAC-d5)、甜蜜素-d11(CYC-d11)、三氯蔗糖-d6(SUC-d6)、泛影酸-d6(DTZ-d6)、碘帕醇-d8(IOD-d8)、碘普罗胺-d3(IOP-d3)、碘美普尔-d3(IOM-d3)、碘海醇-d5(IOX-d5)均购于加拿大Toronto Research Chemicals公司,咖啡因-d3(CAF-d3)购于美国o2si公司。甲醇(色谱纯,德国Merck公司);甲酸、乙酸铵(色谱纯,德国CNW公司);离子对试剂Tris(色谱纯,美国Sigma-Aldrich公司);实验用水为Milli-Q水。
HR-X固相萃取柱(500 mg/6 mL,Chromabond公司);HLB固相萃取柱(500 mg/6 mL,Waters公司);玻璃纤维滤膜(GF/F,孔径70 mm,英国 Whatman公司);尼龙过滤器(13 mm × 0.22 μm,上海安谱公司)。
1.2 标准溶液的配制
所有标样和内标均溶于甲醇中配成单标储备液,各标样的浓度均为100 mg/L,各内标的浓度均为10 mg/L。用甲醇分别配制浓度为10 mg/L的混合标样储备液和浓度为1 mg/L的混合内标储备液。工作液则在使用前由储备液稀释而得。所有储备液均置于-18 ℃ 冰柜中保存备用。
1.3 样品前处理
固相萃取前,水样经70 mm GF/F滤膜过滤,以去除水样中的颗粒物及悬浮物。然后准确量取一定体积的水样(污水处理厂进水200 mL;出水500 mL;地表水500 mL),向其中加入混合内标(使水样中各内标质量浓度均为100 μg/L),摇匀。固相萃取柱依次用10 mL甲醇和10 mL超纯水(4 mol/L硫酸调至pH 3.0)活化,水样以5~10 mL/min的流速通过固相萃取柱。水样加载完后,在真空条件下抽干柱子2 ~3 h。以8 mL甲醇洗脱,洗脱液氮吹近干,以甲醇定容至1 mL,过0.22 μm有机相尼龙滤膜,在-18 ℃下保存。仪器分析前,取出100 μL样品,吹干后用相应模式的缓冲液重新定容,采用UPLC-MS/MS进行检测。
1.4 色谱-质谱条件
目标化合物采用液相色谱-串联质谱进行测定,离子源为电喷雾(ESI)离子源,其中人造甜味剂在ESI(-)模式下分析,造影剂和咖啡因在ESI(+)模式下分析,扫描方式为多反应监测(MRM)模式。色谱柱为Zorbax SB-C18(100 mm×3 mm,1.8 μm),柱温:40 ℃,流速:0.3 mL/min,进样量:20 μL。
ESI(+)模式下流动相:A为水(10 mmol/L乙酸铵,0.01%甲酸),B为乙腈,梯度洗脱程序:0~ 6 min,2% B;6~8 min,2%~ 60% B;8~13 min,60% B;13~18 min,60%~2% B。干燥气温度:350 ℃;干燥气流量:10.0 mL/min;雾化器压力:206.9 kPa;电喷雾电压:200 V。ESI(-)模式下流动相:A为水(5 mmol/L乙酸铵,1 mmol/L Tris),B为乙腈。梯度洗脱程序:0~3 min,5% B;3~5 min,5%~ 90% B;5~16 min,90%~5% B。干燥气温度:350 ℃;干燥气流量:6.0 mL/min;雾化器压力:344.8 kPa;电喷雾电压:200 V。
使用Opitimizer软件(美国Agilent公司)对各单标及内标进行扫描,选择丰度最高的两组母离子/子离子对分别作为定量和定性离子并记录其对应的碎裂电压(Fragmentor)和碰撞能(Collision energy)。各目标化合物及内标的质谱采集参数见表1。

表1 目标化合物的UPLC-MS/MS优化质谱参数Table 1 Mass transitions and retention times for target compounds in UPLC-MS/MS
*quantitative ion;I.S.:internal standard
2 结果与讨论
2.1 仪器条件的优化
流动相是影响峰形、响应值、分离度、柱压和分析时间的重要因素。本文分别以甲醇和乙腈作为有机相进行对比,发现乙腈作为有机相时,各物质的峰形较好。ESI(+)模式下,在流动相中加入缓冲盐和弱酸可以调节流动相的pH值,并为目标化合物提供质子,从而提高化合物的响应值,对流动相中乙酸铵和甲酸的浓度进行优化,确定了最优的流动相为乙腈-水(10 mmol/L乙酸铵,0.01 %甲酸);在ESI(-)模式下,流动相中加入离子对试剂Tris可以显著提高各人造甜味剂的响应值[17],本文对流动相中Tris和乙酸铵的浓度进行优化,得到最优的流动相为乙腈-水(5 mmol/L乙酸铵,1 mmol/L Tris)。在优化的质谱与色谱条件下,目标化合物的总离子流图见图1。


图1 混合标准溶液(100 μg/L)的总离子流图Fig.1 Total ion chromatograms(TIC) of mixed standard solution(100 μg/L)

图2 不同pH值下目标化合物的绝对回收率Fig.2 Absolute recoveries of target compounds under various pH values
2.2 样品前处理条件的优化
2.2.1 固相萃取小柱的选择 以超纯水为基质进行加标回收实验,对HLB和HR-X两种固相萃取柱的萃取效率进行考察。由于本文采用同位素内标对回收率进行校正,因此两种固相萃取柱对各目标化合物均有较好的相对回收率(82%~125%)。通过绝对回收率对两种柱进行对比,结果发现,除ASP和SUC外,HR-X柱对其他10种化合物的绝对回收率均高于HLB柱,因此本文采用HR-X柱进行固相萃取。
2.2.2 样品pH值的选择 样品的pH值对萃取效率有较大的影响,因此本文考察了不同pH值(3.0,5.0,7.0)下各目标化合物的萃取效率。结果显示,各目标化合物在3个pH值条件下的相对回收率均在70%~120%之间,但在pH 7.0时绝对回收率均较低,pH 3.0时,甜味剂ACE、SAC、CYC、ASP和离子型造影剂ITA、DTZ有较高的绝对回收率,尤其是ASP在pH 3.0条件下的绝对回收率显著提高(图2),因此实验选择样品的pH值为3.0。
2.3 线性范围与相关系数
分别用正、负模式的缓冲液配制5,10,50,100,200,500,1 000,2 000,5 000 μg/L系列浓度的造影剂和咖啡因的混合标准溶液以及人造甜味剂的混合标准溶液,内标浓度均为100 μg/L。在优化条件下,采用内标法,以待测物的峰面积与对应内标的峰面积的比值(Y)为纵坐标,质量浓度(X,μg/L)为横坐标绘制标准工作曲线。结果显示,各目标化合物在5~5 000 μg/L范围内线性关系良好,相关系数(r2)为0.998 8~0.999 9。
2.4 精密度、准确度、检出限与定量下限
采用优化的方法对地表水、污水处理厂出水和进水进行加标回收实验,由于不同类型的水样中各目标化合物的本底值差别较大,因此,各类化合物在不同水样中的加标浓度不同。目标化合物在地表水、污水处理厂出水和进水中的回收率分别为70.1%~117%、72.9%~117%和71.7%~119%,相对标准偏差(RSD)均小于20%。基于UPLC-MS/MS对最低加标浓度的样品进行分析得到信噪比,以3倍信噪比对应的浓度为方法检出限(MDL),10倍信噪比对应的浓度为定量下限(MQL)。由表2~4可知,地表水、污水处理厂出水和进水中MDL分别为0.43~13.8、1.80~34.8、2.30~134 ng/L,MQL分别为1.45~45.8、6.02~116、7.67~445 ng/L。

表2 地表水中各目标化合物的回收率(R)、基质效应、方法检出限与定量下限Table 2 Recoveries,matrix effects,method detection limits(MDLs) and method quantification limits(MQLs) of target compounds from surface water
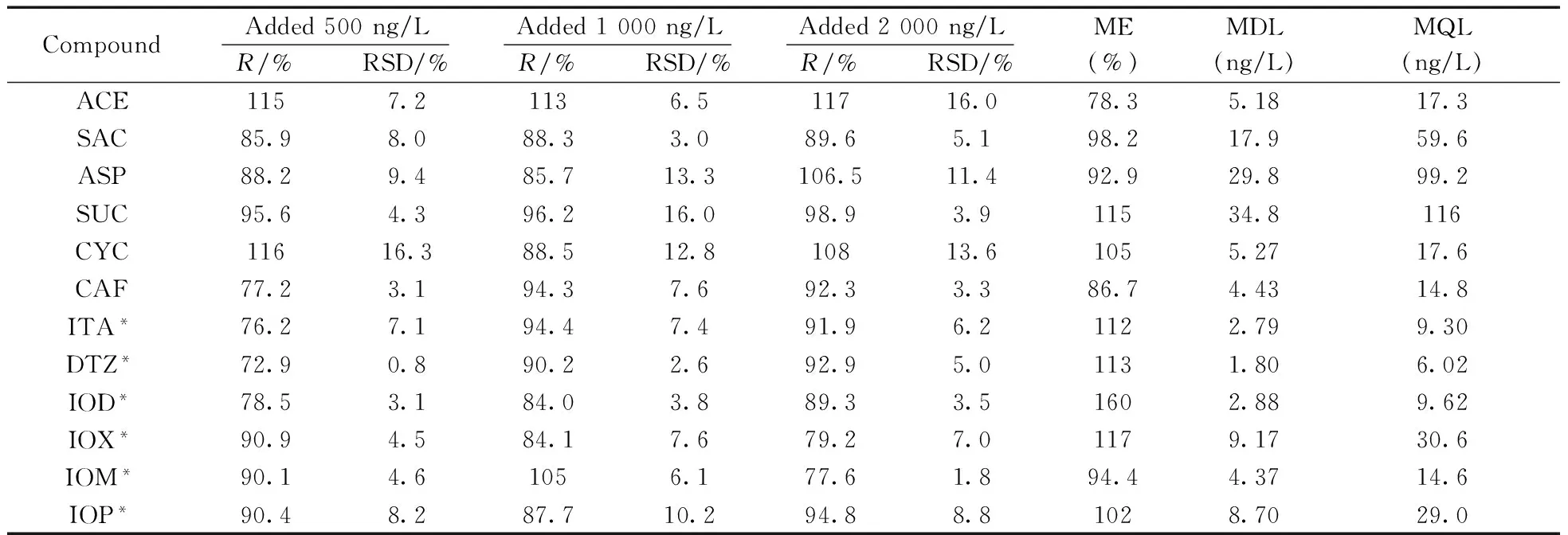
表3 污水处理厂出水中各目标化合物的回收率、基质效应、方法检出限与定量下限Table 3 Recoveries,matrix effects,MDLs and MQLs of target compounds from effluent
*indicated that the spiked concentrations were 100,500,1000 ng/L,respectively

表4 污水处理厂进水中各目标化合物的回收率、基质效应、方法检出限与定量下限Table 4 Recoveries,matrix effects,MDLs and MQLs of target compounds from influent
*indicated that the spiked concentrations were 100,500,1000 ng/L,respectively
此外,本文还考察了目标化合物在不同类型水样中的基质效应(ME),计算公式为ME=[(CS-CN)/CSp-sol]×100%。其中CS为相应基质提取液中添加标准溶液后各目标化合物的浓度;CN为相应基质提取液中各目标化合物的浓度;CSp-sol为起始流动相中添加相应浓度的标准溶液后各目标化合物的浓度。结果显示,目标化合物在地表水(除了CYC,65%)、污水处理厂进水(除了SUC为124%)和出水(除了IOD为160%)中未受到明显的基质干扰(70%~120%)。
2.5 实际样品的测定
采用本方法对广州某污水处理厂的进出水及受纳河流上下游的地表水进行分析检测。结果表明,ASP,ITA,IOM和IOP在各水样中均未检出,其余8种化合物在所有水样中普遍存在且浓度较高,其中ACE,SAC,SUC和CAF的浓度达μg/L级(见表5)。

表5 不同水样中各目标化合物的浓度Table 5 Concentrations of target compounds in different water samples (ng/L)
*no detected
3 结 论
本文建立了检测不同水体(地表水、污水处理厂进水和出水)中5种人造甜味剂、6种造影剂及咖啡因的UPLC-MS/MS方法,该法具有灵敏度高、重现性好、基质干扰小等优点,为全面普查人造甜味剂、造影剂及咖啡因在地表水、污水处理厂进水和出水中的含量水平及其分布特征提供了基础数据。
[1] Buerge I J,Buser H R,Kahle M,Muller M D,Poiger T.Environ.Sci.Technol.,2009,43(12):4381-4385.
[2] Tran N H,Hu J,Li J,Ong S L.WaterRes.,2014,48(1):443-456.
[3] Wolf L,Zwiener C,Zemann M.Sci.TotalEnviron.,2012,430(15):8-19.
[4] Buerge I J,Poiger T,Müller M D,Buser H R.Environ.Sci.Technol.,2003,37(4):691-700.
[5] Buerge I J,Poiger T,Müller M D,Buser H R.Environ.Sci.Technol.,2006,40(13):4096-4102.
[6] Zhu Y,Guo Y Y,Ye M L,James F S.J.Chromatogr.A,2005,1085(1):143-146.
[7] Ferrer I,Thurman E M.J.Chromatogr.A,2010,1217(25):4127-4134.
[8] Liu X X,Ding L,Liu J X,Zhang Y,Huang Z Q,Wang L B,Chen B.Chin.J.Chromatogr.(刘晓霞,丁利,刘锦霞,张莹,黄志强,王立兵,陈波. 色谱),2010,28(11):1020-1025.
[9] Liu L M,Peng J D.J.Instrum.Anal.(刘丽敏,彭敬东. 分析测试学报),2008,27(5):549-552.
[10] Ordonez E Y,Quintana J B,Rodil R,Cela R.J.Chromatogr.A,2012,1256(21):197-205.
[11] Gan Z,Sun H,Wang R,Feng B.J.Chromatogr.A,2013,1274(25):87-96.
[12] Sacher F,Raue B,Brauch H.J.Chromatogr.A,2005,1085(1):117-123.
[13] Echeverria S,Borrull F,Fontanals N,Pocurull E.Talanta,2013,116:931-936.
[14] Dong L L,Yu L,Li M,Wang L C.J.Instrum.Anal.(董丽丽,于玲,李敏,王立川. 分析测试学报),2013,32(6):742-746.
[15] Verenitch S S,Lowe C J,Mazumder A.J.Chromatogr.A,2006,1116(1 /2):193-203.
[16] Hillebrand O,Nodler K,Licha T,Stuter M,Geyer T.WaterRes.,2012,4(2):395-402.
[17] Scheurer M,Brauch H J,Lange F T.Anal.Bioanal.Chem.,2009,394(6):1585-1594.
Rapid Determination of Five Artificial Sweeteners,Six Iodinated X-ray Contrast Agents and Caffeine in Different Water Samples by Solid Phase Extraction with Ultra Performance Liquid Chromatography-Tandem Mass Spectrometry
YANG Yuan-yuan1,2,YING Guang-guo1*,ZHAO Jian-liang1,LIU You-sheng1,LIU Wang-rong1
(1.State Key Laboratory of Organic Geochemistry,Guangzhou Institute of Geochemistry,Chinese Academy of Sciences, Guangzhou 510640,China;2.The University of Chinese Academy of Science,Beijing 100049,China)
An ultra performance liquid chromatography-tandem mass spectrometric(UPLC-MS/MS) method was developed for the rapid determination of 5 artificial sweeteners,6 iodinated X-ray contrast agents and caffeinein in various water samples(surface water,inflow and effluent for wastewater treatment plant). The filtered water samples were extracted,and purified through HR-X solid phase extraction cartridges and separated on a Zorbax SB-C18column. The determination of analytes was performed under multiple reaction monitoring(MRM) mode using acetonitrile-water(10 mmol/L ammonium acetate,0.01% formic acid) and acetonitrile-water(5 mmol/L ammonium acetate,1 mmol/L Tris) as mobile phases in positive ionization mode and negative ionization mode,respectively. The calibration curves of the target compounds were linear in the range of 5-5 000 μg/L with correlation coefficients more than 0.998. The recoveries of the target compounds in surface water,inflow and effluent were all between 70% and 120%,with relative standard deviations(RSD) of 0.8%-19.0%. The method detection limits(MDL) for three types of water samples were in the ranges of 0.43-13.8 ng/L,1.80 -34.8 ng/L and 2.30-134 ng/L,respectively. The method was successfully applied in the determination of artificial sweeteners,iodinated X-ray contrast agents and caffeine in different actual water samples collected from a wastewater treatment plant and its receiving river from Guangzhou city. Except for aspartame,iothalamic acid,iopromide and iomeprol,the other compounds were all detected with high concentrations. The established method is accurate,sensitive and simple,and is suitable for the determination of artificial sweeteners,iodinated X-ray contrast agents and caffeine in various environmental water samples.
artificial sweeteners;iodinated X-ray contrast agents;caffeine;ultra performance liquid chromatography-tandem mass spectrometry(UPLC-MS/MS);water samples
2016-06-21;
2017-01-13
东江流域饮用水源型河流水质安全保障技术集成与综合示范(2014ZX07206-005);国家自然科学基金项目(U1401235)
10.3969/j.issn.1004-4957.2017.07.006
O657.63;O741.6
A
1004-4957(2017)07-0876-06
*通讯作者:应光国,博士,研究员,研究方向:环境化学与生态毒理学,Tel:020-85290200,E-mail:guang-guo.ying@gig.ac.cn;guangguo.ying@gmail.com
猜你喜欢
现代仪器与医疗(2021年4期)2021-11-05 08:25:28
中华养生保健(2020年4期)2020-11-16 01:30:44
大自然探索(2017年10期)2017-10-28 06:47:59
大自然探索(2017年5期)2017-05-26 17:48:07
实用临床医学(2016年8期)2016-06-07 01:28:16
现代养生·上半月(2016年8期)2016-05-14 09:09:24
少年科学(2015年10期)2015-10-31 04:19:47
医学研究杂志(2015年7期)2015-06-22 11:01:01
江苏调味副食品(2015年1期)2015-02-28 01:56:34
中国医学科学院学报(2014年6期)2014-03-11 20:26:19
