
反门控去耦技术在定量1H NMR测定苯乙醇胺A纯度中的应用
2017-08-01 12:46:35宋印清王彤彤杨梦瑞
分析测试学报 2017年7期
宋印清,王 敏,王彤彤,周 剑,杨梦瑞
(中国农业科学院 农业质量标准与检测技术研究所,北京 100081)
反门控去耦技术在定量1H NMR测定苯乙醇胺A纯度中的应用
宋印清,王 敏*,王彤彤*,周 剑,杨梦瑞
(中国农业科学院 农业质量标准与检测技术研究所,北京 100081)
采用定量核磁中反门控去耦的方法对苯乙醇胺A的纯度进行测定。通过对氘代溶剂和内标物、定量峰的选择,以及弛豫延迟时间(D1),采样次数和采样温度等定量核磁条件的优化,最终确定测试条件为:激发脉冲角度30,时间域数据点32 K,测定温度293 K,脉冲的弛豫延迟为33.64 s,采样次数64,线宽0.3 Hz。在此实验条件下,稳定性可达24 h,耐用性良好。以样品和内标的峰面积比对其摩尔比绘制标准曲线,结果显示,两者的摩尔比在0.333~3.333范围内线性关系良好,相关系数(r2)为0.999 9。该方法专属性强,准确,简便,适用于该类型化合物含量的测定。
苯乙醇胺A;定量核磁;反门控去耦;纯度
苯乙醇胺A又名克伦巴胺,是一种β-受体激动剂,具有β-受体激动剂的典型结构。它是一种人工合成的化学物质,是福莫特罗的同分异构体,其结构(见图1)与莱克多巴胺类似[1]。苯乙醇胺A是2010年新发现的一种瘦肉精,具有营养再分配的作用,添加到饲料中可加速脂肪的分解和转化,提高动物的瘦肉率、减少饲料使用,降低成本[2];但人食用上述物质会产生心律失常、心慌、心悸、甲状腺机能亢进等中毒症状[3],可诱发和加重心脑血管病人的病情,因此我国颁布了1519号公告,禁止将苯乙醇胺A等11种物质添加于饲料和动物饮用水中。国内外针对苯乙醇胺A的测定主要以高效液相色谱和液相色谱-质谱联用方法为主。
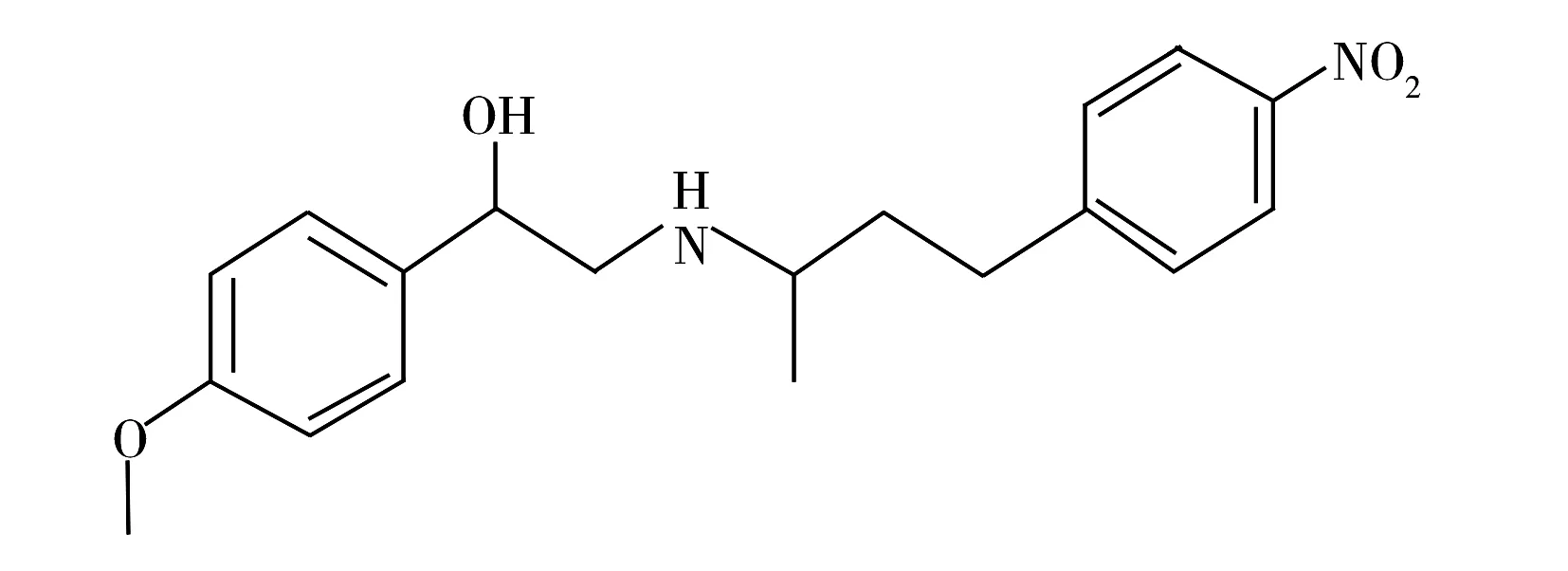
图1 苯乙醇胺A的结构式 Fig.1 Structural formula of phenylethanolamine A
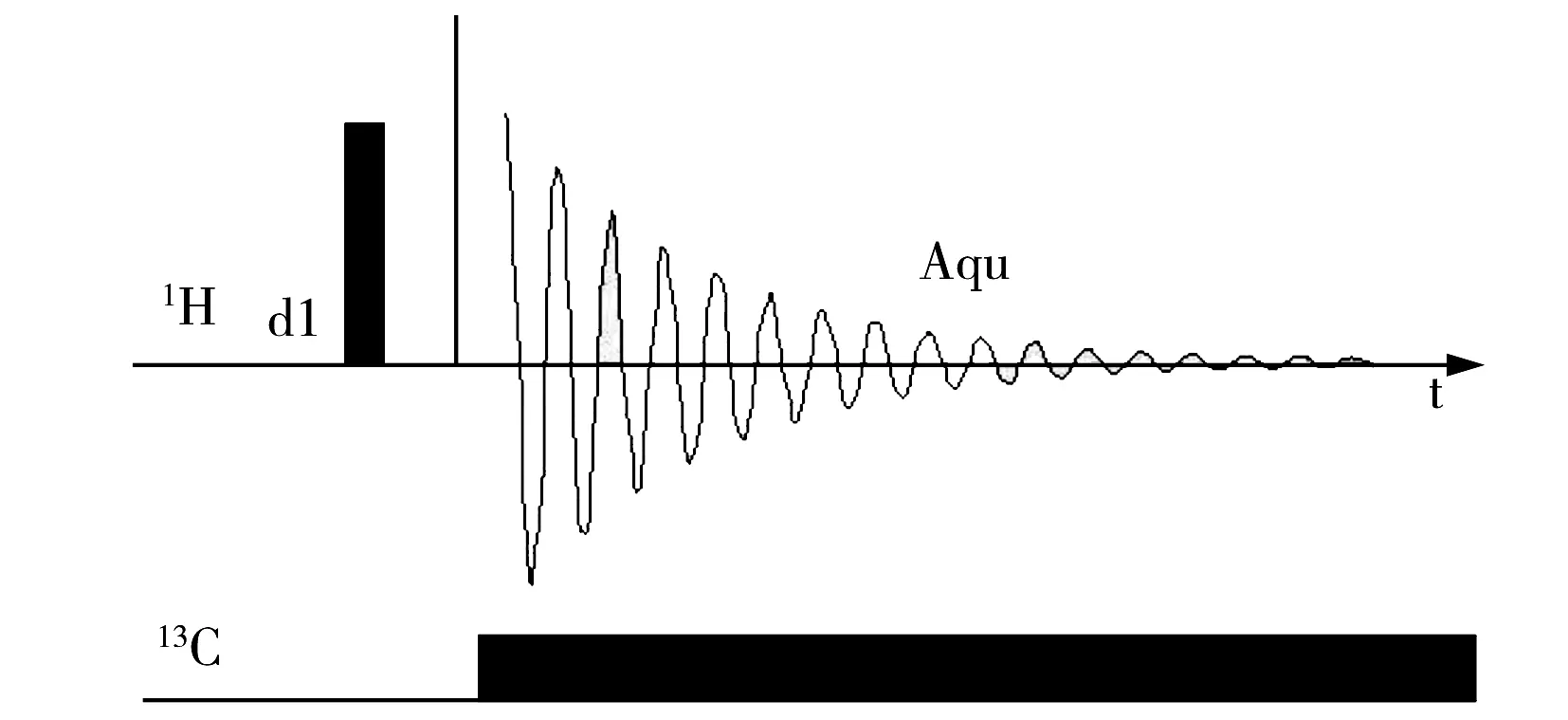
图2 氢谱中反转门控去耦的脉冲序列Fig.2 Pulse sequence for inverse gated decoupling in 1H NMR
定量核磁共振(Quantitative nuclear magnetic resonance,qNMR)不需要待测物对照品,仅以已知含量的药物或化学物质为参比即可对待测物含量进行测定[4]。该方法具有前处理步骤简单、快捷等优点,已被各国药典所收录[5-6]。近年来,qNMR技术已被广泛用于定量分析,主要包括药物的含量测定[7-8]、异构体[9]和食品领域[10]。qNMR包括1H,13C,19F,31P,14N,15N-NMR等,其中1H由于丰度和灵敏度高,适用范围广,因此被国内外研究机构广泛报道[11-13]。核磁共振中常规的去耦方式[14]包括宽带去耦、门控去耦和反门控去耦。宽带去耦无耦合,有奥弗豪塞尔核效应(NOE);门控去耦既有耦合现象也有NOE现象,对于氢谱和碳谱定量和解析均有明显的影响;反门控去耦技术是指去耦器在弛豫延迟期关闭而在采样期打开,脉冲序列如图2所示,该技术既无耦合现象也无NOE现象,最适于氢谱和碳谱的定量。本文在定量氢谱中运用反门控去耦技术测定了违禁兽药苯乙醇胺A的纯度,并用于苯乙醇胺A纯度标准物质的制作。
1 实验部分
1.1 仪器与试剂
Bruker AVANCE Ⅲ型400 MHz核磁共振仪(瑞士Bruker公司);UMX2型电子天平(分度值0.1 μg,瑞士梅特勒公司);苯乙醇胺A(Laboratorien Berlin Adlerhof GmbH,德国);氘代甲醇(纯度99.8%,美国Sigma公司);尼泊金乙酯(中国计量科学研究院,GBW(E)100064), 5 mm核磁管(WG-1000-7, 美国Wilmad公司)。苯甲酸 (中国计量科学研究院,GBW(E)100064),邻苯二甲酸氢钾 (中国计量科学研究院,GBW(E)100064)。
1.2 实验方法
1.2.1 样品配制方法 分别精确称量约1.0~10.0 mg样品和3.0 mg内标物各3份置于同一棕色玻璃瓶,用1.0 mL氘代甲醇振荡完全溶解后,转移至直径为5 mm的核磁试管中,待测。
1.2.2 核磁实验参数与图谱处理方法1H NMR测定参数:进行3D匀场和自动调谐,激发脉冲角度30,采集时间4.0 s,时间域数据点64 K,扫描宽度20 ppm, 弛豫延迟 (D1≥5T1),自动调节增益, 中心频率(位于定量峰和内标峰的中心位置), 窗函数0.3 Hz。
图谱的处理方法:通过Topspin 2.1软件进行数据采集,利用ACD Lab软件进行处理,均依次采用自动和手动相位调整,自动和手动基线校准及根据氘代甲醇的氢谱数据进行化学位移校正。在基线与峰的重合扩大± 0.1 Hz范围内进行积分。
1.2.3 计算方法 取苯乙醇胺A和尼泊金乙酯,按样品制备方法平行配制3份样品,样品振荡溶解后,在优化条件下记录图谱,每个样品平行测量5次,并取平均值。
式中:PNMR为采用核磁共振法测得的被测物纯度;Ix和Istd分别为样品和内标物指定峰的积分面积;nx和nstd分别为样品和内标物指定峰的核群核个数;Mx和Mstd分别为样品和内标物的分子量;mx和mstd分别为样品和内标物的质量;Pstd为内标物的纯度。
2 结果与讨论
2.1 氘代溶剂与内标物的选择
氘代溶剂应使样品和内标有良好的溶解度。考虑到苯乙醇胺A的极性大,因此选择氘代甲醇作为溶剂。内标物选择的依据为:易溶于氘代试剂,化学结构稳定,属于国家级标准物质,且不与待测化合物反应。由于两种内标苯甲酸和邻苯二甲酸氢钾均呈酸性,易与苯乙醇胺A发生化学反应,生成季铵盐,因此不适合作为本实验的内标。国家级标准物质尼泊金乙酯不与苯乙醇胺A发生化学反应,易溶于氘代甲醇中,且定量峰分布均匀,与苯乙醇胺A均含有苯环结构,因此采用尼泊金乙酯作为内标。
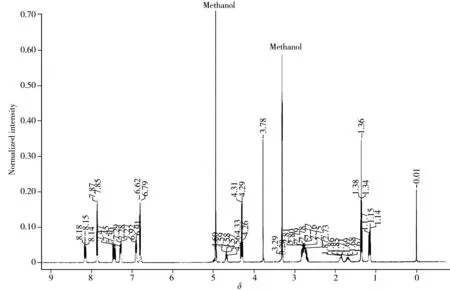
图3 苯乙醇胺A和尼泊金乙酯混合物的氢谱图Fig.3 qNMR spectra of phenylethanolamine A and ethyl paraben mixture
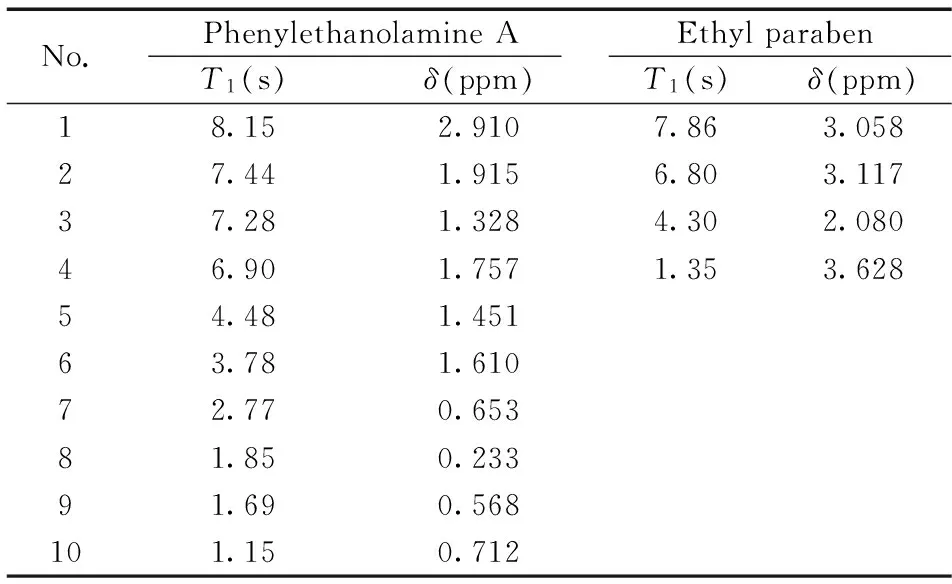
No.PhenylethanolamineAEthylparabenT1(s)δ(ppm)T1(s)δ(ppm)18.152.9107.863.05827.441.9156.803.11737.281.3284.302.08046.901.7571.353.62854.481.45163.781.61072.770.65381.850.23391.690.568101.150.712
determination longitudinal relaxation delay time(T1):(T1 delay=0.01,0.05,0.1,0.25,0.5,1,2,4,8,15μs)were measured by inversion recovery method,and the T1values of different peaks were obtained by data fitting(纵向弛豫时间T1测定方法:采用反转恢复法测定T1,利用软件Topspin 2.1进行采样(T1delay=0.01, 0.05,0.1, 0.25, 0.5, 1, 2, 4, 8, 15 μs), 经过数据拟合得到不同峰的T1值)
2.2 氢谱定量峰的选择
定量峰必须满足在积分范围中无干扰,且信号的化学位移和纵向弛豫时间(T1)尽可能接近内标物的信号峰,以减少误差。在两者混合物氢谱中发现(如图3),苯乙醇胺A的高场区可能受到微量杂质的影响,而苯环区基线平稳,受到杂质影响的几率较小。同时苯乙醇胺A的化学位移在δH8.15处共振谱线的纵向弛豫时间为2.910 s,而内标物尼泊金乙酯在δH7.86处纵向弛豫时间为3.058 s,两者之间十分接近,且两峰之间的化学位移相差大于0.4 Hz,满足qNMR对内标和检测物标定谱线选择的条件 (表1)。确定两定量峰后,将定量氢谱的中心频率设在δH8.00 处,以降低偏共振效应(Off-resonance effect)的影响。
2.3 核磁条件的优化
2.3.1 脉冲弛豫延迟时间(D1)的选择 根据文献报道[15],D1/T1比值增加,会提高实验的精确度。为了保证定量峰的自旋核能够达到99%以上弛豫,必须使D1/T1≥5,因此需进一步优化两者比值,以满足定量核磁的要求。在优化的定量峰条件下,纵向弛豫时间T1=3.058 s,分别考察不同D1/T1(5、8、11、14、17)条件下样品与内标物的定量峰面积比,其结果分别为1.723、1.725、1.727、1.727、1.727。D1/T1= 11时,样品与内标物的定量峰面积比值无明显变化,能够满足定量要求,因此选择D1= 33.64 s。
2.3.2 采样次数的选择 取1.0 mg苯乙醇胺A和3.0 mg尼泊金乙酯内标,D1= 33.64 s,在不同采样次数(8、16、32、64、128次)条件下,分别测定定量峰8.15 ppm与基线9.0 ppm间的信号强度,利用Topspin 2.1软件计算S/N。结果表明,谱图的信噪比S/N与采样次数的平方根成正比,扩大采样次数有利于提高采样的信噪比。由于核磁共振定量要求S/N≥250,因此确定采样次数为64次。
2.3.3 采样温度的选择 取约1.0 mg苯乙醇胺A和3.0 mg尼泊金乙酯内标,在上述优化条件下,对采样温度(T=15、20、25、30、35 ℃)进行优化。实验数据表明采样温度对内标物和样品的积分面积影响很小,可以忽略不计。
2.4 方法学验证
2.4.1 线性范围 分别精密称取样品约1.0、2.0、4.0、6.0、10.0 mg,内标物尼泊金乙酯3.0 mg,置于同一离心管中,按照“1.2”的方法平行配制5份样品溶液,按照“2.3”优化后的条件测量氢谱。以样品/内标摩尔比为横坐标(x),样品/内标定量峰面积比为纵坐标(y)进行线性拟合。结果表明,样品与内标的摩尔比在0.333 3~3.333 3范围内,氢信号与样品量成正比,方法的线性关系良好,回归方程为y=1.013 7x-0.001 6,相关系数(r2)为0.999 9。
2.4.2 精密度 精密称取4.0 mg苯乙醇胺A和3.0 mg尼泊金乙酯,按照“2.3”优化后的条件测量日内和日间的氢谱和碳谱。结果显示,定量氢谱中苯乙醇胺A与尼泊金乙酯峰面积(NA/NA)的日内相对标准偏差(RSD)为0.15%,日间RSD为0.18%。相对于其他去耦方式,反门控去耦[16]可以造成13C对1H的耦合作用,因此该方法的精密度更适合于定量氢谱。
2.4.3 方法比较 利用“2.4.2”制备在核磁管中的样品,与常见定量氢谱进行方法比对,结果显示:常见定量氢谱采用宽带去耦方式,其日内相对标准偏差为0.35%,高于反门控去耦的日内相对标准偏差(0.15%),原因可能是受到卫星峰的影响。
2.5 方法的应用
取购于国外公司的两份苯乙醇胺A标准品,分别进行精确称量,并加内标物尼泊金乙酯于试管中,完全溶解后注入核磁管中,在上述优化条件下进行测定,分别测得两种样品中苯乙醇胺A的含量为99.3%(RSD=0.18%)和99.5%(RSD=0.11%),表明该方法结果准确、可靠。
3 结 论
通过反门控去耦消除卫星峰,有利于谱线积分区域的设定,在谱线相对比较接近时可抑制卫星峰信号对标定峰定量积分的影响。因此,定量核磁方法中的反门控去耦技术有望成为定量方法,相信随着该技术在分析领域应用的不断深入,能为不同领域成分和含量分析开拓新的思路和方法。
[1] Sun W Y,Zhao B L,Zhang S J,Sun Z L,Zhang L.Chin.J.Chromatogr.(孙武勇, 赵冰琳, 张守杰, 孙转莲, 张莉.色谱), 2012,30(10):1008-1011.
[2] Eshaq (Isaac) S, Sin C C, Sami J, Gene A, Michael K H.Anal.Chim.Acta, 2003,483:137-145.
[3] Zhang Y,Zhao S Z,Qu L,Cao C,Shi Y Y,Yi X H.FoodSci.( 张旖,赵善贞,曲栗,曹晨,时逸吟,伊雄海.食品科学), 2014,35(20):202-207.
[4] Chen X L, Guo Y J, Hu Y J, Yu B Y, Qi J.J.Pharm.Biomed.Anal., 2016,124:281-287.
[5] Holzgrabe U, Deubner R, Schollmayer C, Waible B.J.Pharm.Biomed.Anal., 2005,38(5):806-812.
[6] National Pharmacopoeia Committee.The Pharmacopoeia of People′s Republic of China (Part Ⅱ,2010Ed.).Beijing:China Medical Science Press.
[7] Sharma R,Gupta P K,Mazumder A,Dubey D K,Ganesan K,Vijayaraghavan R.J.Pharm.Biomed.Anal., 2009,49(4):1092-1096.
[8] Chauthe S K, Sharma R J,Aqil F,Gupta R C,Singh I P.Phytochem.Anal.,2012,23(6):689-696.
[9] Li J,Geng Z F,Liu P,Deng Z W.Chin.Chem.Lett.,2012,23(10) :1181-1184.
[10] del Campo G,Berregi I,Caracena R,Zuriarrain J.Talanta,2010,81(1/2):367 -371.
[11] Manoukian P, Melliou E , Liouni M, Magiatis P.Lwt-FoodSci.Technol., 2016,65:1133-1137.
[12] Kuchta K, Ortwein J, Hennig L,Rauwald H W.Fitoterapia,2014,96:8-17.
[13] Wahl O, Holzgrabe U.J.Pharm.Biomed.Anal., 2014,95:1-10.
[14] Guido F P, Birgit U J, David C L.J.Nat.Prod.,2007,70:589-595.
[15] Bharti S K, Roy R.TrendAnal.Chem., 2012,35:5-26.
[16] Timothy D W C.High-ResolutionNMRTechniquesinOrganicChemistry.2nd ed.Singapore:Elsevier, 2010.
Application of Inverse Gated Decoupling Technique in Quantitative1H NMR Determination of Purity of Phenylethanolamine A
SONG Yin-qing,WANG Min*,WANG Tong-tong*,ZHOU Jian,YANG Meng-rui
(Institute of Quality Standard and Testing Technology for Agro-products,Chinese Academy of Agriculture,Beijing 100081,China)
A high accuracy method of qNMR with inverse gated decoupling technique was established to determine the content of phenylethanolamine A.Considering the choice of deuterated solvents,internal standards and quantitative peaks,and the optimization of relaxation delay,aquisition number and temperature,the final measurement conditions of qNMR spectra were as follows:flip angle:30,date point:32 K,detection temperature:293 K,a relaxation delay:33.64 s,acquisition times:64 times,line broadening:0.3 Hz.The experimental results showed that the stability of this condition could be maintained for 24 h.A good linear relationship was obtained between peak area ratio and molar ratio in the range of 0.333-3.333,and the correlation coefficient (r2) was 0.999 9.The established method is specific,accurate and simple,and could be used for the content determination of this kind of compounds.
phenylethanolamine A;qNMR;inverse gated decoupling;purity
2016-12-22;
2017-03-11
中国农业科学院创新工程;农产品质量安全风险评估标准物质研制与质量控制规范研究(2015)(GJFP201501503)
10.3969/j.issn.1004-4957.2017.07.018
O482.532;TQ460.72
A
1004-4957(2017)07-0933-04
*通讯作者:王 敏,研究员,研究方向:标准与标准物质,Tel:010-82106546,E-mail:wangmincaas@126.com 王彤彤,博士,助理研究员,研究方向:有机合成,Tel:010-82106545,E-mail:wangttong123@126.com
猜你喜欢
云南化工(2023年7期)2023-08-01 07:59:34
现代仪器与医疗(2022年4期)2022-10-08 05:54:40
现代临床医学(2022年4期)2022-09-29 07:36:10
中国抗生素杂志(2022年7期)2022-08-18 03:22:36
农产品加工(2021年8期)2021-05-20 01:38:28
大学化学(2021年2期)2021-04-09 11:15:32
长春师范大学学报(2019年4期)2019-04-29 05:51:36
百姓生活(2016年6期)2016-06-22 14:39:00
中国高新技术企业(2015年13期)2015-04-30 21:07:38
实验室研究与探索(2015年3期)2015-02-21 06:26:55
