
柱前衍生化—HPLC检测亚氨基二乙酸法制备双甘膦反应进程的研究
2017-07-05 01:15:24庄勇黄楷
安徽农业科学 2017年14期
关键词:高效液相色谱
庄勇 黄楷
摘要[目的]建立一种利用亚氨基二乙酸制备双甘膦的新检测方法。[方法]以9-芴基甲氧羰酰氯为衍生化试剂对合成双甘膦的反应物进行衍生化。采用Gemini C18 (150.0 mm ×4.6 mm, 5.0 μm)色谱柱,室温下以0.05 mol/L NaH2PO4 (pH=3.5)溶液-乙腈(V/V,60∶40)为流动相,流速为1.0 mL/min,检测波长265 nm,对反应液中的亚氨基二乙腈、亚氨基二乙酸和氨气衍生物进行分离检测。[结果]反应液中的亚氨基二乙腈、亚氨基二乙酸和氨气衍生物,分别在0.02~1.20、0.02~1.20、0.10~2.00 mg/mL浓度范围内呈现良好的线性关系(r≥0.999 8),检出限分别为0.000 4、0.000 4、0.003 0 mg/mL,加样回收率分别为99.96%、99.86%、99.89%,相对标准偏差均不超过1.6%。[结论]该研究建立的方法可用于亚氨基二乙酸法制备双甘膦反应体系的跟踪分析。
关键词亚氨基二乙酸法;双甘膦;高效液相色谱;衍生化
中圖分类号O657.7文献标识码A文章编号0517-6611(2017)14-0012-02
Abstract[Objective] To establish a new inspection method for preparation of PMIDA with iminodiacetic acid.[Method] The derivatives of the reaction solution for synthesizing PMIDA were synthesized using 9fluorenyl methyl chloroformate (FMOCCl) as the derivatization reagent.The separation of the derivatives was performed on a Gemini C18 column at room temperature by isocratic elution.The mobile phase composed of 60% 0.05 mol/L NaH2PO4 (pH=3.5) and 40% CH3CN at a flow rate of 1.0 mL/min.The injection volume was 10 μL and the detection wavelength was 265 nm.[Result] The linear ranges of the derivatives of iminodiacetonitrile, iminodiacetic acid and ammonia were respectively 0.02-1.20 mg/mL, 0.02-1.20 mg/mL and 0.10-2.00 mg/mL with correlation coefficient of more than or equal to 0.999 8.The detection limits for the derivatives were respectively 0.000 4, 0.000 4 and 0.003 0 mg/mL.The average recoveries of 3 derivatives were respectively 99.96%, 99.86% and 99.89% with relative standard deviation values less than or equal to 1.6%.[Conclusion] The method developed in the present paper could be used for trace analysis of the reaction system for preparation of PMIDA by iminodiacetic acid.
Key wordsIminodiacetic acid;PMIDA;HPLC;Derivatization
草甘膦是美國孟山都公司于1974 年商品化的灭生性有机膦类除草剂[1],是目前世界农药市场上吨位最大、销售额最高的除草剂品种。双甘膦是制备草甘膦的重要中间体[2-3],其生产过程中涉及的物质包括亚氨基二乙腈、亚氨基二乙酸和氨气等。在实际生产过程中需要一个准确的分析检测方法对整个工艺流程进行中间控制,了解原料以及中间体的反应消耗情况,才能对整个生产工艺过程做出正确的分析和判断。为此,建立一种能快速准确监控双甘膦制备进程的分析方法就显得尤为重要。
由于该反应过程中涉及的含氮化合物的结构中均缺少发色团,难以采用常规的高效液相色谱法(HPLC)直接测定。为此,笔者根据高效液相色谱中脂肪胺类化合物的柱前衍生原理[4],以荧光试剂9-芴基甲氧羰酰氯(FMOC-Cl)[5-7]与反应液中的胺类化合物柱前衍生,然后用HPLC进行分离测定。
1材料与方法
1.1材料Shimadzu LC-2010型高效液相色谱仪,日本岛津;FMOC-Cl(>99%),Sigma-Aldrich;亚氨基二乙腈(纯度≥98%),广州江城石化;氢氧化钠(含量≥98%)、盐酸(含量35%),国药集团;亚磷酸(含量89%),上海实诚;甲醛(含量37%),上海凌风;乙腈(色谱纯),北京百灵威;超纯水,自制。
1.2方法
1.2.1标准品衍生方法。称取亚氨基二乙腈、亚氨基二乙酸、合成中间体等标准样品约25.0 mg置于50 mL容量瓶中,加20 mL硼酸缓冲液(0.2 mol/L、pH 7.5),再加20 mL FMOC-Cl乙腈溶液(0.4 mmol),用乙腈定容至刻度,在室温25 ℃下放置10 min,配制成0.5 mg/mL浓度的待测标准样品溶液。
1.2.2样品衍生化方法。亚氨基二乙酸中控样品配制:称取反应液体约2 mL(约400 mg)置于烧杯中,用3 mol/L盐酸水溶液调节pH至7±1,并用纯水稀释至约100 mL,再取0.2 mL该溶液,加入0.8 mL硼酸缓冲液和0.8 mL FMOC-Cl乙腈溶液(0.016 mmol),配制成约0.8 mg/mL的待测样品溶液,静置10 min待測。
双甘膦中控样品衍生化配制:称取反应液体约2 mL(约400 mg)置于烧杯中,用28%氢氧化钠水溶液调节pH至7±1,并用纯水稀释至约100 mL,再取0.2 mL该溶液,加入0.8 mL硼酸缓冲液和0.8 mL FMOC-Cl/乙腈溶液(0.016 mmol),配制成约0.8 mg/mL的待测样品溶液,静置10 min待测。
1.2.3色谱分离条件。Gemini C18色谱柱(5.0 μm,4.6 mm×150.0 mm);流动相A为0.05 mol/L NaH2PO4 (pH=3.5),流动相B为 乙腈,A∶B=60%∶40%(V/V);流速0.8 mL/min;进样量8 μL;柱温35 ℃;波长265 nm。
2结果与分析
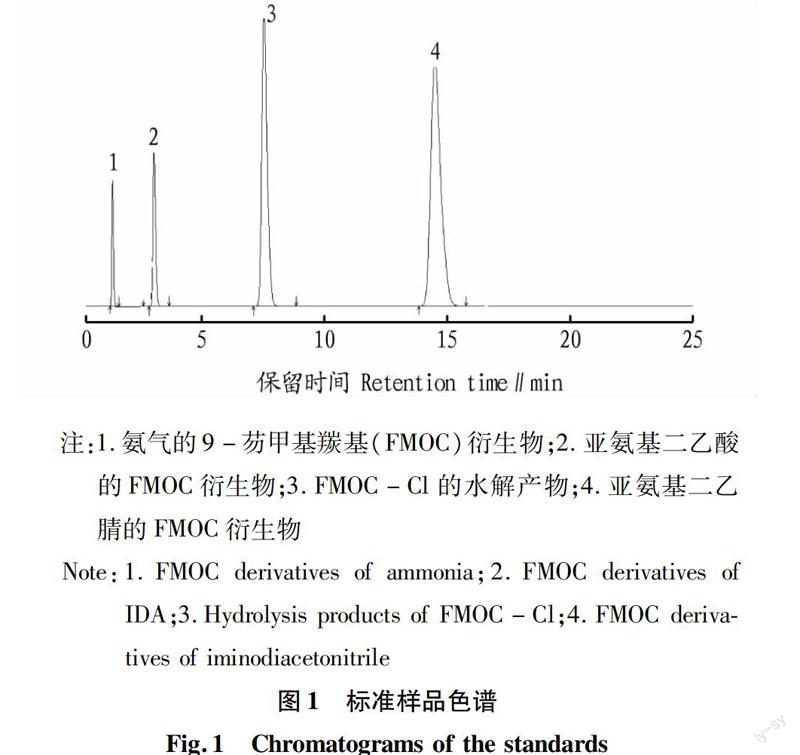
2.1分离条件的优化该试验分别选择了不同浓度的磷酸二氢钠-磷酸缓冲溶液作为流动相对亚氨基二乙腈、亚氨基二乙酸以及氨的衍生物同时检测,发现只有当流动相中磷酸二氢钠的浓度为0.05 mol/L,用磷酸将体系的pH调到3.5时最合适,原料和中间体出峰相对较快,峰形对称性好,且各物质的分离效果较好,各标准品的色谱图见图1。
2.2衍生方法的重现性按照上述衍生化方法,对亚氨基二乙腈、亚氨基二乙酸、氨气分别平行衍生5次,然后采用HPLC进行分析。结果表明,衍生物保留时间的RSD分别为0.38%、0.62%、0.52%,峰面积的RSD分别为1.60%、1.20%、1.10%。表明该衍生化方法重复性良好。
2.3衍生产物的稳定性将上述样品衍生反应液在室温下保存3 d后测定,结果表明,各衍生产物的峰型、峰面積和保留时间等均无明显差异,各组分的衍生产物检测结果的RSD均小于2%,表明各组分的衍生产物稳定性良好。
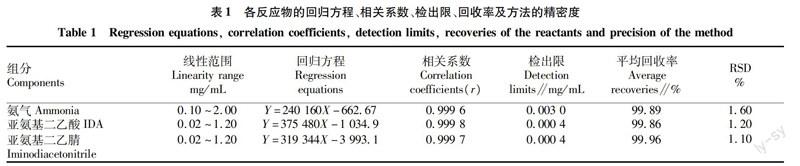
2.4标准曲线、准确度、精密度和检出限配制一系列标准溶液,按方法“1.2.1”衍生和“1.2.3”进行色谱分析。以各组分的质量浓度为横坐标,峰面积为纵坐标,进行线性回归,以3倍信噪比计算检出限,结果如表1所示。在一定的浓度范围内,各组分峰面积和浓度呈良好的线性关系,相关系数均在0.999 5以上。精密量取已测定含量的反应溶液,分别加入高、中、低3种不同浓度的标准溶液,混匀,衍生化后进样分析,考察分析方法的加样回收率和RSD。结果表明,加样回收率分别为99.89%、99.86%、99.96%,RSD分别为1.60%、1.20%、1.10%。
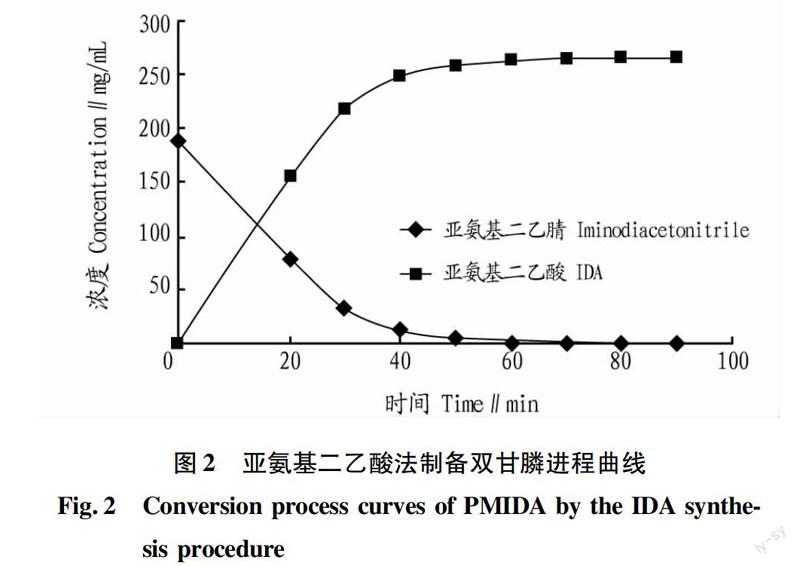
2.5实际样品反应过程分析称取一定的反应原料,在小于45 ℃投料时即产生大量的氨气,同时迅速生成亚氨基二乙酸进行反应,按照一定的时间间隔取适当的反应液,按照“1.2.2”和“1.2.3”的方法进行样品处理和分析,测定体系中各组分的浓度,考察反应过程中底物和产物的变化,确定最适合的反应条件。如图2所示,随着反应的进行,原料亚氨基二乙腈的浓度下降,亚氨基二乙酸浓度逐渐升高,在60 min
时达到最大。所以建立的跟踪监测方法对亚氨基二乙酸法制备双甘膦反应有很大帮助。
3结论
该试验建立的方法具有简便快速、灵敏准确、衍生条件温和及衍生产物稳定性好等特点,能对亚氨基二乙酸法制备双甘膦的工业化生产进程进行准确的跟踪和检测,对降低该路线的工业化生产成本和提高产品质量具有重要意义。
参考文献
[1] 苏少泉.草甘膦述评[J].农药,2005,44(4):145-149.
[2] 张海斌,张小宏,王建清.降低双甘膦生产能耗改进措施[J].化工中间体,2007(5):26-27.
[3] 张海斌,张小宏,尹岳明,等.三氯化磷替代亚磷酸合成双甘膦工艺改进[J].现代农药,2007,6(5):19-21.
[4] 张琳,平贵臣,于晓明,等.氯甲酸-9-芴甲酯柱前衍生脂肪胺类化合物的反相高效液相色谱分析[J].现代仪器,2004,10(2):14-16.
[5]EINARSSON S,JOSEFSSON B,LAGERKVIST S.Determination of amino acids with 9fluorenylmethyl chloroformate and reversedphase highperformance liquid chromatography[J].Journal of chromatography A,1983,282:609-618.
[6] ANSON MOYE H,BONING A J JR.A versatile fluorogenic labelling reagent for primary and secondary amines:9fluorenylmethyl chloroformate[J].Analytical letters,1979,12(1):25-35.
[7] BAUZA T,BLAISE A,DAUMAS F,et al.Determination of biogenic amines and their precursor amino acids in wines of the Vallée du Rhone by highperformance liquid chromatography with precolumn derivatization and fluorimetric detection[J].Journal of chromatogr A,1995,707:373-379.
猜你喜欢
分析化学(2017年1期)2017-02-06 21:32:17
中国医药导报(2016年30期)2016-12-28 12:18:02
科技创新与应用(2016年33期)2016-12-17 13:35:35
科技创新与应用(2016年33期)2016-12-17 13:34:28
热带农业科学(2016年10期)2016-12-12 01:52:56
分析化学(2016年7期)2016-12-08 00:57:07
上海医药(2016年21期)2016-11-21 23:14:07
中国科技博览(2016年18期)2016-10-19 11:09:28
科学与财富(2016年28期)2016-10-14 04:01:52
中国科技博览(2016年2期)2016-04-25 14:06:58
