
结构域对小麦蛋白质二硫键异构酶性质的影响*
2017-05-30 02:34胡松青黄政刘光黄滟波李琳侯轶
华南理工大学学报(自然科学版) 2017年11期
胡松青 黄政 刘光,2 黄滟波 李琳 侯轶
(1.华南理工大学 食品科学与工程学院, 广东 广州 510640; 2.广东省农业科学院 蚕业与农产品加工研究所,广东 广州 510610; 3.华南理工大学 轻工科学与工程学院, 广东 广州 510640)
蛋白质二硫键异构酶(PDI)是位于真核细胞内质网中的一种多功能酶,它主要由4个硫氧还蛋白结构域按照a-b-b′-a′顺序排列组成,并在C-末端含有一段由大量酸性氨基酸组成的c尾[1].结构域a和a′含有催化活性位点CXXC基序(C为半胱氨酸,X为除半胱氨酸以外的氨基酸),非催化结构域b和b′虽不含活性位点,但存在与底物结合的疏水表面.正是这种结构特性,使得PDI在催化新生肽链巯基/二硫键交换反应中发挥着重要作用,其能够催化二硫键的氧化、还原以及异构反应[2].除了催化巯基/二硫键交换反应,PDI还有另一个重要功能——分子伴侣,PDI的分子伴侣活性表现为促进新生或者变性蛋白质的折叠以及恢复蛋白质的天然活性[3].
PDI每个结构域对其活性贡献不同,通过删除或突变PDI结构域可以考察PDI各结构域和活性位点对其活性的影响[4].发挥巯基/二硫键交换反应的最小单元为a或a′结构域,其活性大小与突变全长PDI另一个结构域活性位点的突变体活性相当[5].Klappa等[6]通过考察人来源PDI(hPDI)截短蛋白对多肽底物的结合作用发现,hPDI的所有结构域都对底物结合发挥作用,且b′结构域提供了最主要的结合位点.另外,Tian等[7]通过截除hPDI的c尾考察了截短蛋白对变性乳酸脱氢酶的分子伴侣活性大小,发现hPDI的c尾有助于稳定PDI的分子伴侣活性.
目前,人和酵母来源PDI的性质和结构得到了较为深入的研究[8- 9],而有关植物来源PDI的性质研究较为鲜见.相比于动物和微生物来源,植物来源PDI表现出不同的功能性质,如大豆PDI包含非典型的氧化还原基序CXXC/S,其在体外不具有氧化还原活性和分子伴侣活性[10].因此,有关植物来源PDI的功能性质研究有待深入.
笔者所在课题组前期利用大肠杆菌原核表达了具有生物活性的重组小麦蛋白质二硫键异构酶(wPDI)[11],文中以wPDI为研究对象,通过表达wPDI各结构域线性组合的截短蛋白,探究了其结构域和活性位点氧化还原状态对wPDI活性的贡献,以期为深入理解wPDI的功能性质奠定基础.
1 材料与方法
1.1 材料与试剂
1.1.1 宿主菌株与质粒
克隆菌株E.coliDH5α、表达菌株E.coliBL21(DE3)购于宝生物工程有限公司;pET- 30b-wpdi重组表达质粒由笔者所在实验室自行构建并保存.
1.1.2 宿主菌株与质粒
主要试剂:胰岛素、牛核糖核酸酶(RNase)、胞苷2′,3′-环单钠磷酸盐(2′,3′-cCMP)和三磷酸甘油醛脱氢酶(GAPDH),购自美国西格玛试剂公司;限制性内切酶BamHI、XhoI和T4 DNA连接酶,购自赛默飞世尔科技(中国)有限公司;DNA凝胶回收试剂盒和DNA小提试剂盒,购自广州英赞生物有限公司;还原型谷胱甘肽(GSH)、氧化型谷胱甘肽(GSSG)和丙基硫代半乳糖苷(IPTG)等试剂均为分析纯,购自健阳生物科技有限公司.
主要仪器:EDC- 80型基因扩增仪,东胜创新生物科技有限公司生产;Infinite M200 Pro型酶标仪,瑞士TECAN公司生产;蛋白电泳仪,美国Bio-Rad公司生产;NanoVue plus型微量紫外分光光度计、VCX500型超声破碎仪,美国SONICS公司生产.
1.2 分析及测试方法
1.2.1 wPDI截短蛋白的构建分析
从小麦中克隆得到的wpdi基因(genbank No.AF262979.1)全长1 548 bp,与“Wyuna”品种小麦PDI基因同源性达99%.wpdi基因编码515个氨基酸,去除信号肽序列后剩余490个氨基酸,按照Johnson等[12]的报道,各结构域的界限分别为:a结构域包含17-120位氨基酸,b结构域包含128-221位氨基酸,b′结构域包含244-338位氨基酸,a′结构域包含358-459位氨基酸.按照结构域界限对wPDI进行线性组合排列,设计出8个截短蛋白片段,分别为ABB′A′、BB′A′C、ABB′、B′A′C、AB、BB′、A′和A.
1.2.2 wPDI各截短蛋白的引物设计
不同wPDI截短蛋白的PCR扩增引物是以wpdi基因序列为模板,利用Primer Piemier 3.0设计的,具体信息见表1.
1.2.3 wPDI重组表达载体的构建
以pET- 30b-wpdi质粒为模板,分别加入wPDI各截短片段上、下游引物,进行PCR扩增反应,反应条件如下:94 ℃预变性3 min后,按以下的参数进行35次循环——98 ℃变性10 s,60 ℃退火15 s,72 ℃延伸90 s.最后,72 ℃延伸5 min后,利用琼脂糖凝胶电泳检测扩增产物,并用DNA凝胶回收试剂盒回收各片段基因.
对基因回收产物和表达载体pET- 30b(+)分别进行BamHI和XhoI双限制性内切酶处理,酶切反应条件为37 ℃、30 min.反应产物回收、连接后转化至大肠杆菌DH5α感受态细胞中,并涂布在LB固体平板培养基上培养.菌落长成后,随机挑选单克隆菌落培养,提取质粒进行限制性内切酶BamHI和XhoI双酶切鉴定,酶切反应条件同上.将经双酶切和电泳验证结果为阳性的重组表达质粒送样测序,测序结果与已知的wpdi基因序列对应结构域部分进行比对,确认重组质粒序列信息.
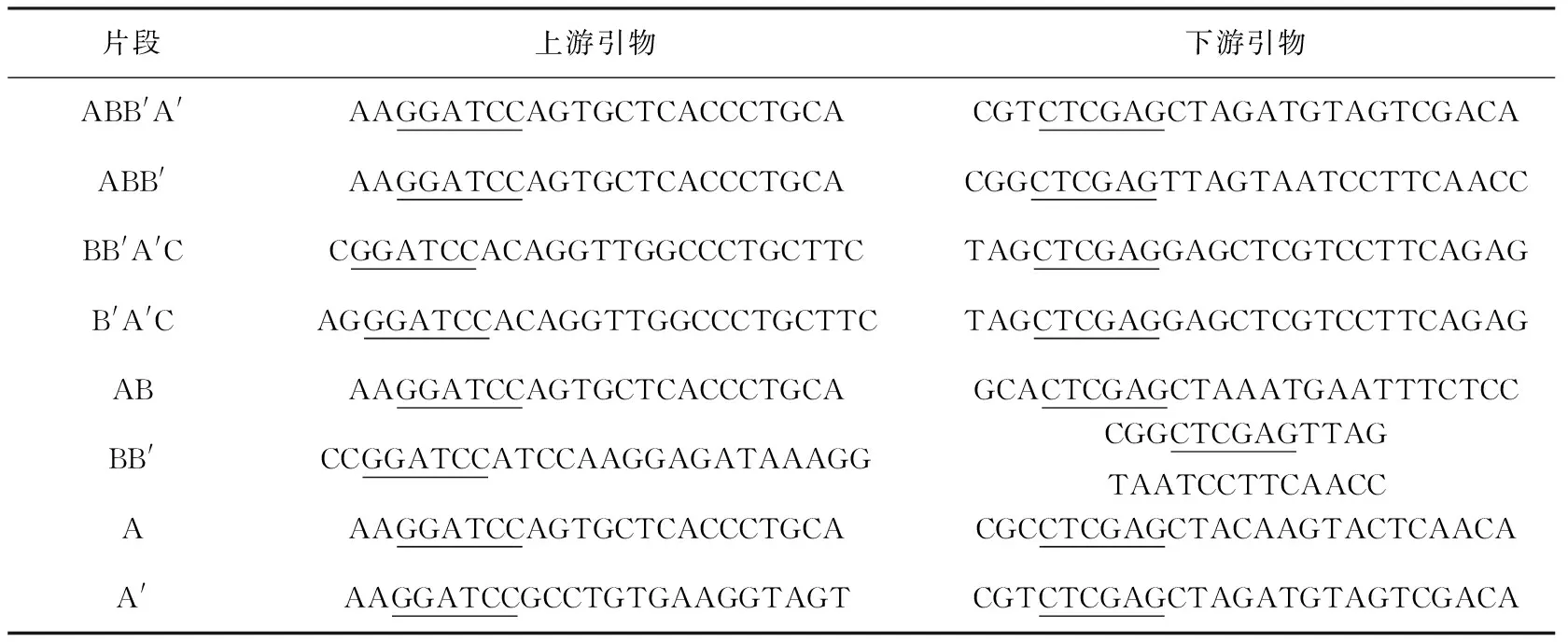
表1 不同wPDI截短蛋白的引物设计1)
1.2.4 表达与纯化
将测序正确的各重组表达质粒分别转化至BL21(DE3)中,并涂布在含Kan的LB固体平板培养基上.37 ℃培养过夜后,挑取形态饱满的单克隆菌接种到含有Kan的LB液体培养基中,继续培养8~10 h后,将菌液以1∶100(体积比)的比例转接到新的LB液体培养基中,相同条件下培养2~3 h至菌液600 nm吸光值达到0.5~0.6后,加入IPTG诱导重组蛋白质的表达.ABB′A′、ABB′、BB′A′C、B′A′C和AB等截短蛋白添加终浓度为0.5 mmol/L的IPTG进行诱导,BB′、A 和 A′截短蛋白添加终浓度为0.1 mmol/L的IPTG诱导.各截短蛋白的诱导表达条件均为20 ℃、200 r/min下诱导过夜.利用SDS-PAGE检测重组蛋白质的表达.
使用Ni-NTA金属螯合层析和分子排阻层析组合层析方法对wPDI各截短蛋白进行分离纯化.纯化后的蛋白保存于含0.15 mol/L NaCl的20 mmol/L Tris-HCl缓冲液(pH=8.0)中,不含有对蛋白结构和活性有影响的SDS和巯基还原剂.各截短蛋白的蛋白质浓度采用微量紫外分光光度计测定,不同截短蛋白的消光系数ε(mL·mg-1·cm-1)值分别为0.805(ABB′A′)、0.745(ABB′)、0.661(BB′A′C)、0.787(B′A′C)、0.576(AB)、0.606(BB′)、0.817(A′)和0.790(A),计算方法参考Simonian等[13]的报道.
1.2.5 活性测定
wPDI包含多种酶学活性,包括二硫键氧化酶、还原酶以及异构酶活性[11].此外,wPDI还具有分子伴侣功能.不同活性的测定方法和底物制备方法参考曹佩等[14]的报道.
二硫键氧化酶和异构酶的活性定义如下:单位时间内生成1 μmol 3′-CMP所需酶量为一个酶活力单位,文中选取反应时间10 min和20 min内的线性区域分别计算各截短蛋白质的二硫键氧化酶和异构酶活性.通过计算开始沉淀时(吸光值相对提高0.01个单位)的延迟时间来分析不同截短蛋白质二硫键还原活性的大小.分子伴侣活性的定义如下:30 min内抑制吸光值增加0.01个单位所需酶量为一个酶活力单位.
1.2.6 蛋白质电泳
使用还原型十二烷基硫酸钠聚丙烯酰胺凝胶电泳(SDS-PAGE)检测截短蛋白质的纯度,使用非还原型SDS-PAGE检测截短蛋白A和A′活性中心的氧化还原状态.还原型和非还原型SDS-PAGE方法参照文献[15]中的方法.
2 结果与讨论
2.1 wPDI截短蛋白的表达
按照wPDI的各结构域界限,对其按线性排列方式进行分段组合,设计出8个截短片段,各片段的结构域组成以及氨基酸个数如图1所示.
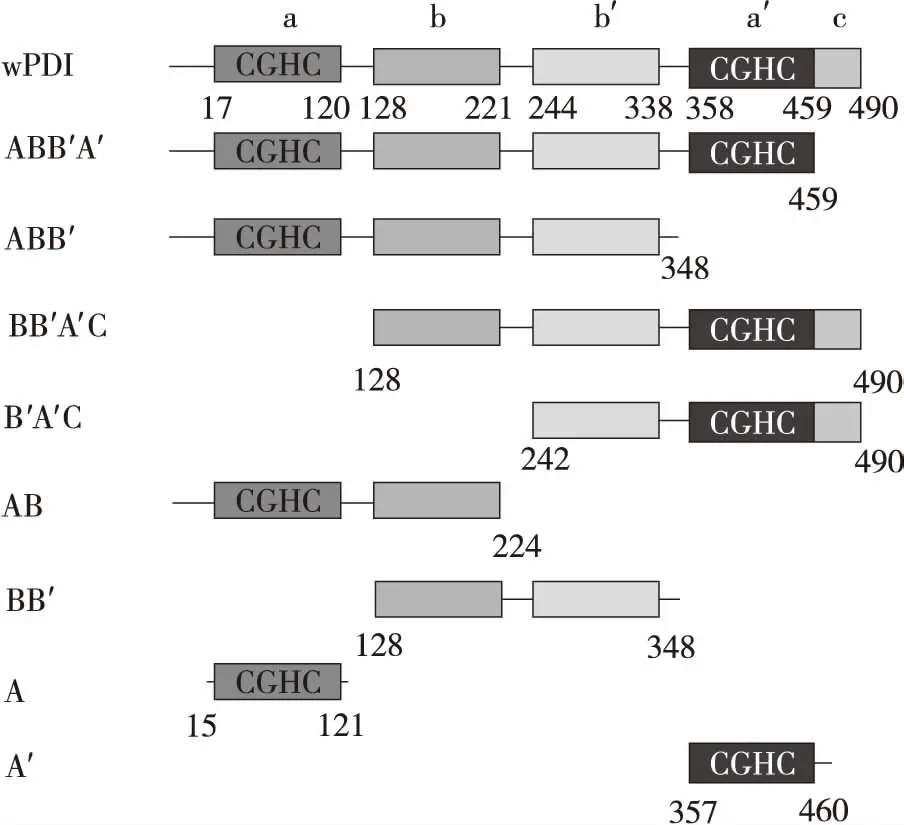
图1 wPDI各截短蛋白的结构域信息
Fig.1 Structure domain information of truncated proteins of wPDI
利用亚克隆方法构建不同截短蛋白重组表达载体.将构建好的各重组表达质粒分别转化到大肠杆菌BL21(DE3)中,诱导表达后利用SDS-PAGE进行表达分析,结果如图2(a)所示.由图可见:截短蛋白ABB′A′、BB′A′C、ABB′、B′A′C、BB′以及A′在电泳胶上的条带十分明显,说明它们具有可观的表达水平;而截短蛋白AB以及A在电泳胶上的条带较淡,说明它们虽有表达,但表达量相对于其他截短蛋白低.
2.2 wPDI截短蛋白的纯化
各截短蛋白的N-端都带有6×His融合标签,利用Ni-NTA金属螯合亲和层析以及分子排阻层析组合层析方法对各截短蛋白进行纯化,纯化后利用还原型SDS-PAGE检测各蛋白质纯度,结果如图2(b)所示.由图可见,wPDI的各截短蛋白在电泳胶上主要以单一条带存在,它们在凝胶上的相对分子质量都略大于其理论相对分子质量(含标签),造成各截短蛋白质在凝胶上迁移率变慢的原因可能与重组蛋白所带His标签上碱性氨基酸过多有关[16].另外,尽管各截短蛋白质在电泳图谱上表现为单体蛋白,但由分子排阻分析可得,截短蛋白A、A′和B′A′C应处于聚集态形式,其他截短蛋白为单体形式,蛋白的聚集对其活性可能产生影响.
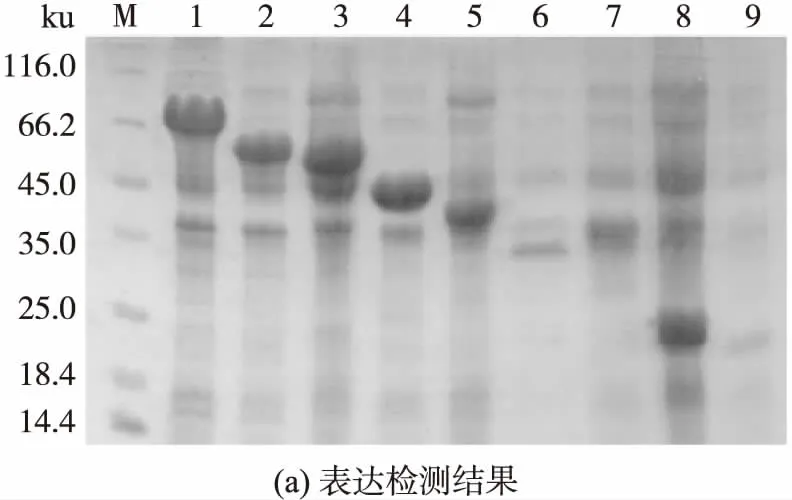
图2 wPDI各截短蛋白的表达检测和纯度分析
2.3 wPDI截短蛋白的活性测定
wPDI包含二硫键的氧化还原活性、异构活性以及分子伴侣活性.因BB′不含催化位点,故仅对其分子伴侣活性进行测定,而对剩下的截短蛋白进行酶学活性和分子伴侣活性的考察,结果如图3所示.
2.3.1 二硫键还原酶活性
图3(a)为不同截短蛋白二硫键还原酶活性测定结果.由图可见,在所选蛋白质中,除B′A′C、A′和A外,在文中实验条件下,其他截短蛋白都表现出了二硫键还原酶活性,其中wPDI表现出了最强的二硫键还原酶活性,说明wPDI所有结构域以及c尾都对其还原胰岛素具有相应的贡献,该结果与Sun等[17]报道的结果一致.
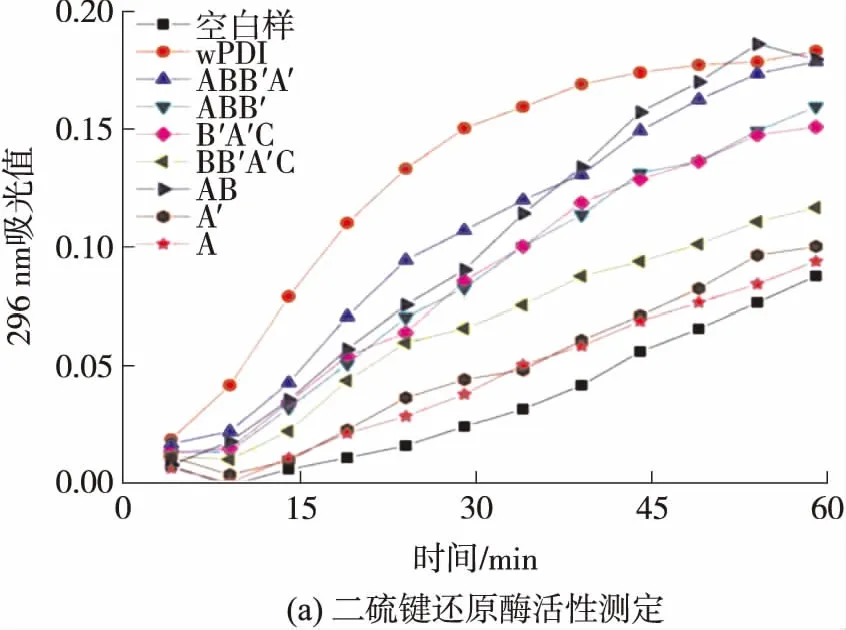
图3 wPDI各截短蛋白的酶学活性和分子伴侣活性测定结果
Fig.3 Results of enzymatic activities and molecular chaperone activity assay of different truncated proteins of wPDI
根据图3(a),以wPDI组与对照组开始沉淀时的时间差值为标准,分别计算各截短蛋白二硫键还原酶活性的相对大小,结果如表2所示.由表可见,仅含有a结构域的截短蛋白的还原活性较不含a结构域的小,如ABB′和AB的二硫键还原酶活性分别为全长wPDI二硫键还原酶活性的8.6%和15.1%,BB′A′C的活性则为67.8%.该结果表明wPDI的a′结构域活性位点可能偏向还原态形式,而a结构域偏向氧化态形式,与hPDI催化活性位点的氧化还原状态类似[18].
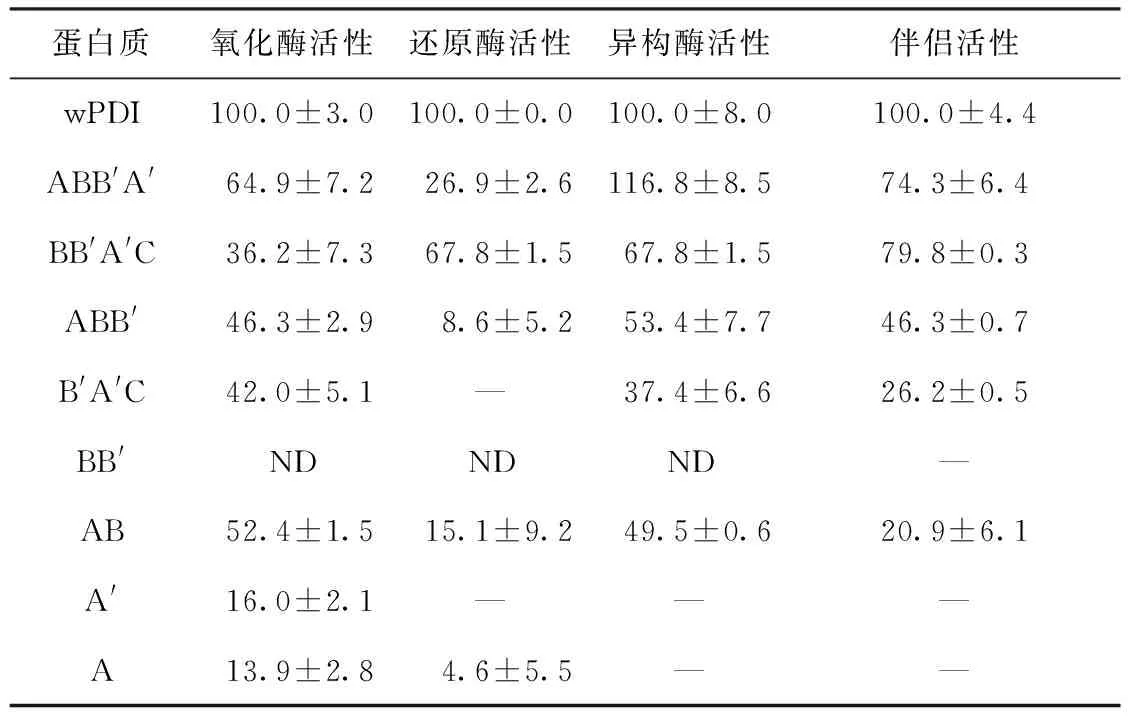
表2 wPDI各截短蛋白的酶学活性和分子伴侣活性比较1)
进一步分析截短蛋白A的电泳结果(见图4)可得,未经DTT(二硫苏糖醇)处理的截短蛋白A单体在电泳图上位置只有一条条带(黑色框标示).添加1 mmol/L DTT后,该单体条带出现了上下两条强度相当并紧挨着的条带.继续增大DTT浓度至2 mmol/L后,电泳图上截短蛋白A单体靠上的条带强度变弱,靠下的条带强度则增强.而当DTT浓度增至4 mmol/L时,靠上的条带基本消失,截短蛋白A单体条带主要以靠下的条带为主.这些结果表明,未加DTT处理的截短蛋白A单体以氧化态形式存在,添加少量DTT(<2 mmol/L)后,截短蛋白A单体以氧化态(上条带)和还原态(下条带)共存.而当DTT浓度进一步增大后,截短蛋白A单体由氧化态完全转变成还原态.该结果与活性测定的结果一致,直接证明了wPDI的a结构域活性位点主要以氧化态形式存在.
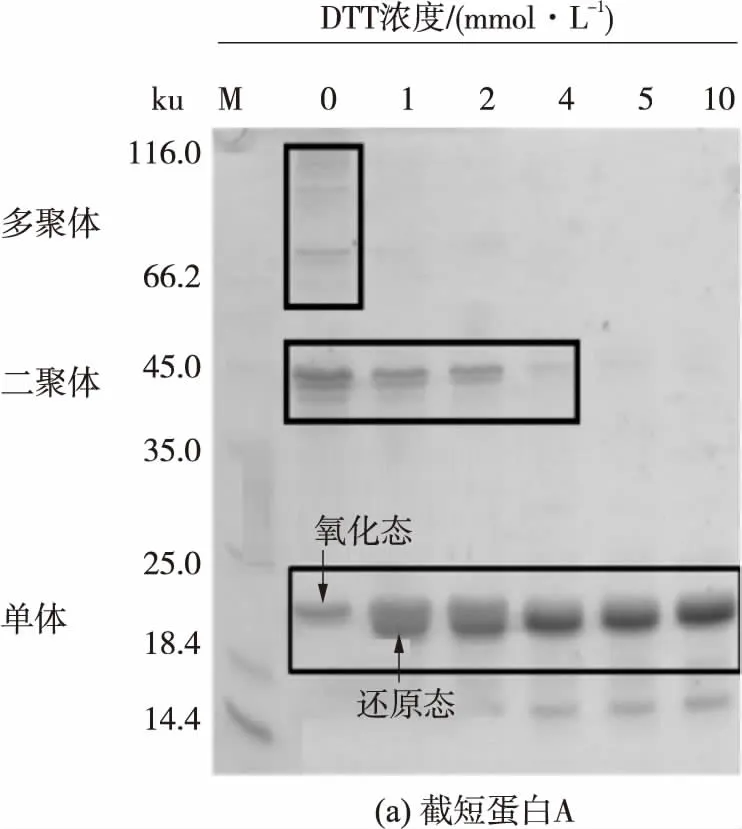
图4 DTT处理下截短蛋白A和A′的非还原SDS-PAGE分析
此外,分析截短蛋白A′的电泳结果发现,在添加DTT处理后,截短蛋白A′单体条带并没有发生与截短蛋白A单体条带相类似的条带迁移率变化(白色框标示),这也从侧面证明了wPDI的a′结构域活性位点主要以还原态形式存在,与活性测定分析的结果相一致.
2.3.2 二硫键氧化酶活性
图3(b)为不同截短蛋白二硫键氧化酶活性测定结果.由图可见,所有截短蛋白都表现出了二硫键氧化酶活性,在30 min内,wPDI引起的溶液吸光值变化最快,说明其二硫键氧化酶活性最高,但随时间的延长,受到底物消耗以及产物抑制作用的影响,wPDI的氧化活性减弱,当时间达到60 min时,ABB′A′和AB实验组引起的溶液吸光值变化量与wPDI组的相近,说明60 min后三者对变性rRNase的复性效率相近.以图3(b)数据计算各截短蛋白的二硫键氧化酶活性相对大小,结果见表2.由表可见,所有截短蛋白的二硫键氧化酶活性都低于全长wPDI,同样说明wPDI的各个结构域对其氧化活性具有一定贡献[4].
此外,仅含a结构域的截短蛋白AB和ABB′的氧化活性要高于仅含a′结构域的截短蛋白BB′A′C和B′A′C,进一步说明wPDI的a结构域活性位点主要以氧化态为主,而a′结构域活性位点主要以还原态为主.另外,截短蛋白A′活性位点主要以还原态为主,故二硫键氧化活性低;而A的活性位点虽然以氧化态为主,但因活性位点半胱氨酸参与形成分子间二硫键交联,导致其氧化活性偏低.
2.3.3 二硫键异构酶活性
图3(c)为不同截短蛋白二硫键异构酶活性测定结果.由图可见,除截短蛋白A和A′外,剩余截短蛋白都表现出了一定的异构活性.研究表明,发挥PDI异构活性的最小结构单元为ab或b′a′c,b′a′c的异构活性与全长PDI突变a结构域活性位点后的突变体异构活性相当[19].比较不同截短蛋白的二硫键异构酶活性发现,截短蛋白ABB′A′的异构活性略高于wPDI,说明c尾并不是异构活性所必需的(如表2所示).此外,截短蛋白所含结构域单元越多,其异构活性也相应越大,同样表明wPDI各结构域对其异构活性都具有贡献,这与Darby等[4]的研究结果相符.
PDI的异构作用包括两种机制[20]:一种是底物分子内直接异构机制,该机制不需要氧化还原剂的参与,仅要求PDI一个活性位点处于还原状态即可;另一种是还原/氧化循环机制,需要两个活性位点和氧化还原当量的同时参与.BB′A′C、B′A′C、ABB′和AB只含有一个催化结构域,因此它们发挥异构活性的机制应为第一种机制;wPDI和ABB′A′含两个催化结构域,因此它们发挥异构作用应同时包含两种机制,这应该是它们的异构活性较其他截短片段大的部分原因.
2.3.4 分子伴侣活性
图3(d)为不同截短蛋白分子伴侣活性测定结果.由图可见,除截短蛋白A、A′和BB′没有表现出分子伴侣活性外,剩余截短蛋白都具有一定的抑制变性GAPDH凝沉的能力.研究表明,PDI的b和b′结构域含疏水区域,提供了与底物结合的关键位点,该结合位点显著影响其异构活性和分子伴侣活性.Horibe等[21]的研究发现,hPDI相关蛋白的a和a′结构域单独存在时不具有分子伴侣活性,这与文中所得结果相似.比较能够发挥分子伴侣活性的截短蛋白发现,各截短蛋白至少需含有一个催化结构域a(或a′)和非催化结构域b(或b′),表明wPDI发挥分子伴侣活性的最小结构单元为ab或b′a′c[21].
表2示出了各截短蛋白对变性GAPDH的相对分子伴侣活性大小.由表可得,截短蛋白所含结构域越多其分子伴侣活性越大,表明每个结构域都有助于PDI结合底物,这与前人所获得的研究结果相符[22].此外,比较wPDI和ABB′A′相对伴侣的活性大小发现,wPDI的c尾截去后损失了25%的全长wPDI分子伴侣活性.Tian等[7]通过比较hPDI和abb′a′(hPDI C-末端29个氨基酸被截除片段)对变性乳酸脱氢酶的分子伴侣活性大小,发现PDI的C-末端有助于稳定PDI的分子伴侣活性.但若hPDI C-末端氨基酸截除数增至51个后,hPDI截短蛋白则丧失了恢复变性GAPDH活性的能力[23- 26].结合这些研究结果分析得到,hPDI的C-末端51个氨基酸中,靠近N端的22个氨基酸包含了对分子伴侣活性至关重要的氨基酸或结合位点,因此,wPDI截短蛋白ABB′A′伴侣活性降低的原因应与C-末端截取的氨基酸有关,截除的41个氨基酸应含与分子伴侣活性相关的关键氨基酸结合位点.
以上结果表明,wPDI的a结构域活性位点主要以氧化态形式存在,a′结构域活性位点主要以还原态形式存在,wPDI的所有结构域对其酶学活性和分子伴侣活性都具有一定的贡献,c尾对其异构活性没有影响,但存在影响分子伴侣活性的关键氨基酸结合位点.
3 结语
文中成功制备了8个wPDI截短蛋白片段,活性试验表明,wPDI所有结构域对其二硫键氧化、还原、异构以及分子伴侣活性都具有贡献,其中,a结构域活性位点主要以氧化态形式存在,a′结构域活性位点主要以还原态形式存在,c尾的缺乏不影响其异构活性,但该部分含有影响分子伴侣活性的关键氨基酸结合位点.研究结果不仅有助于理解wPDI活性与其结构域之间的关系,而且为充分认识不同来源PDI的共性和特性问题起到了促进作用.
:
[1] CREIGHTON T E.Protein disulfide isomerase [J].Biochimica Et Biophysica Acta,2004,1699(1/2):35- 44.
[2] HATAHET F,RUDDOCK L W.Protein disulfide isome-rase:a critical evaluation of its function in disulfide bond formation [J].Antioxid Redox Signal,2009,11(11):2807- 2850.
[3] 王志珍.蛋白质二硫键异构酶既是酶又是分子伴侣 [J].科学通报,1998,43(13):1345- 1353.
WANG Zhi-zhen.Protein disulfide isomerase is a kind of enzyme but also a molecular chaperone [J].Chinese Science Bulletin,1998,43(13):1345- 1353.
[4] DARBY N J,PENKA E,VINCENTELLI R.The multi-domain structure of protein disulfide isomerase is essential for high catalytic efficiency [J].J Mol Biol,1998,276(1):239- 247.
[5] GILBERT H F.Protein disulfide isomerase and assisted protein folding [J].J Biol Chem,1997,272:29399- 29402.
[6] KLAPPA P,RUDDOCK L W,DARBY N J,et al.The b′ domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins [J].Then EMBO Journal,1998,17:927- 935.
[7] TIAN R,LI S,WANG D,et al.The acidic C-terminal domain stabilizes the chaperone function of protein disulfide isomerase [J].The Journal of Biological Chemistry,2004,279:48830- 48835.
[8] TIAN G,KOBER F X,LEWANDROWSKI U,et al.The catalytic activity of protein-disulfide isomerase requires a conformationally flexible molecule [J].The Journal of Bio-logical Chemistry,2008,283:33630- 33640.
[9] TIAN G,XIANG S,NOIVA R,et al.The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites [J].Cell,2006,124(1):61- 73.
[10] IWASAKI K,KAMAUCHI S,WADAHAMA H,et al.Molecular cloning and characterization of soybean protein disulfide isomerase family proteins with nonclassic active center motifs [J].FEBS Journal,2009,276(15):4130- 4141.
[11] 刘光,胡松青,张婷婷,等.小麦蛋白质二硫键异构酶基因的克隆、表达及重组酶性质 [J].食品科学,2017,38(2):7- 14.
LIU Guang,HU Song-qing,ZHANG Ting-ting,et al.Gene cloning,expression and characterization of protein disulfide isomerase from wheat (TriticumaestivumL.) [J].Food Science,2017,38(2):7- 14.
[12] JOHNSON J C,CLARKE B C,BHAVE M.Isolation and characterisation of cDNAs encoding protein disulphide isomerases and cyclophilins in wheat [J].J Cereal Sci,2001,34(2):159- 171.
[13] SIMONIAN M H,SMITH J A.Spectrophotometric and colorimetric determination of protein concentration [M]∥Current Protocols in Molecular Biology.[S.l.]:John Wiley & Sons,2006:10- 11.
[14] 曹佩,卫娜,刘光,等.小麦蛋白质二硫键异构酶的毕赤酵母表达及重组酶性质研究 [J].现代食品科技,2017,33(5):1- 7.
CAO Pei,WEI Na,LIU Guang,et al.Research on the characteristics of recombinant wheat protein disulfide isomerase expressed inPichiapastoris[J].Modern Food Science and Technology,2017,33(5):1- 7.
[15] 刘光毅.重组高分子量麦谷蛋白亚基 1Dx5 的 N 端结构域聚集行为研究 [D].广州:华南理工大学,2015.
[16] 唐威华,张景六,王宗阳,等.SDS-PAGE法测定His-tag融合蛋白分子量产生偏差的原因 [J].植物生理学报,2000,26(1):65- 69.
TANG Wei-hua,ZHANG Jing-liu,WANG Zong-yang,et al.The cause of deviation made in determining the molecular weight of His-tag fusion proteins by SDS-PAGE [J].Acta Phytophysiologica Sinica,2000,26(1):65- 69.
[17] SUN X,DAI Y,LIU H,et al.Contributions of protein disulfide isomerase domains to its chaperone activity [J].Biochimica et Biophysica Acta (BBA)-Protein Structure and Molecular Enzymology,2000,1481(1):45- 54.
[18] ARAKI K,NAGATA K.Functional in vitro analysis of the ERO1 protein and protein-disulfide isomerase pathway [J].J Biol Chem,2011,286:32705- 32712.
[19] XIAO R,SOLOVYOV A,GILBERT H F,et al.Combinations of protein-disulfide isomerase domains show that there is little correlation between isomerase activity and wild-type growth [J].The Journal of Biological Chemistry,2001,276:27975- 27980.
[20] WALKER K W,LYLES M M,GILBERT H F.Catalysis of oxidative protein folding by mutants of protein disulfide isomerase with a single active-site cysteine [J].Bio-chemistry,1996,35(6):1972- 1980.
[21] HORIBE T,IGUCHI D,MASUOKA T,et al.Replacement of domain b of human protein disulfide isomerase-related protein with domain b′ of human protein disulfide isomerase dramatically increases its chaperone activity [J].FEBS Letters,2004,566(1/2/3):311- 315.
[22] PIRNESKOSKI A,KLAPPA P,LOBELL M,et al.Mole-cular characterization of the principal substrate binding site of the ubiquitous folding catalyst protein disulfide isomerase [J].The Journal of Biological Chemistry,2004,279:10374- 10381.
[23] DAI Y,WANG C.A mutant truncated protein disulfide isomerase with no chaperone activity [J].The Journal of Biological Chemistry,1997,272:27572- 27576.
[24] NORGAARD P,WINTHER J R.Mutation of yeast Eug1p CXXS active sites to CXXC results in a dramatic increase in protein disulphide isomerase activity [J].Bio-chemistry Journal,2001,358(1):269- 274.
[25] AKIYAMA Y,KAMITANI S,KUSUKAWA N,et al.In vitro catalysis of oxidative folding of disulfide-bonded proteins by the Escherichia coli dsbA(ppfA)gene pro-duct [J].The Journal of Biological Chemistry,1992,267:22440- 22445.
[26] CAI H,WANG C,TSOU C.Chaperone-like activity of protein disulfide isomerase in the refolding of a protein with no disulfide bonds [J].The Journal of Biological Chemistry,1994,269:24550- 24552.
猜你喜欢
食品科学技术学报(2022年5期)2022-10-11
自然灾害学报(2022年2期)2022-05-10
小学教学研究(2022年5期)2022-04-28
分析化学(2019年4期)2019-05-13
山西地震(2019年1期)2019-03-20
西安交通大学学报(2019年3期)2019-03-08
商周刊(2019年1期)2019-01-31
中国洗涤用品工业(2017年2期)2017-04-16
中国饲料(2016年17期)2016-12-01
通信电源技术(2016年6期)2016-04-20
