
Alagille 综合征合并肝细胞癌1例报告并文献复习
2017-05-25 00:37会杨永臣张泓张婷肖咏梅
临床儿科杂志 2017年4期
胡 会杨永臣张 泓张 婷肖咏梅
上海市儿童医院 上海交通大学附属儿童医院1.消化科, 2.检验科(上海 200062)
Alagille 综合征合并肝细胞癌1例报告并文献复习
胡 会1杨永臣2张 泓2张 婷1肖咏梅1
上海市儿童医院 上海交通大学附属儿童医院1.消化科, 2.检验科(上海 200062)
目的探讨儿童Alagille综合征合并肝细胞癌的临床表现、影像学检查、治疗及预后。方法分析1例Alagille 综合征合并肝细胞癌患儿的临床表现、辅助检查、诊断,并复习相关文献。结果患儿,女,6岁,因反复皮肤黄染入院,曾有心脏手术史。入院后查体有特殊面容(前额突出、眼眶深陷、尖下颌、鼻前端肥大)。血生化提示存在肝内胆汁淤积,甲胎蛋白升高;影像学腹部B超示肝内弥漫性多发实性占位,肝脏磁共振提示肝大,肝脏多发占位。Jagged 1基因检测为c.1205delC,杂合型。结论儿童期Alagllie综合征合并肝细胞癌极为罕见,早期诊断和长期随访对治疗及预后具有积极意义。
Alagille综合征; 肝细胞癌; 儿童
Alagille综合征又称肝动脉发育不良或综合征性小叶间胆管缺乏,以组织学小叶间胆管减少或缺乏为最重要病理特征的一种少见、累及多系统的常染色体显性遗传性疾病,是婴儿期胆汁淤积的重要病因之一。Alagille综合征在1969年由Alagille等首次报道,国外报道发病率约为1/70 000[1]。Alagille综合征涉及到肝脏、心脏、骨骼、眼睛及肾脏的病变,伴有特殊面容。97%的患者因位于染色体20p12的jagged 1基因突变或缺失引起,与Notch 2基因有关的病例不足1%[2]。肝组织活检和分子基因检测为确诊Alagille综合征提供了直接证据。长期肝内胆汁淤积会导致胆汁性肝纤维化。少数关于Alagille综合征合并肝细胞结节性病变的病例已被报道,关于Alagille综合征合并肝细胞癌国内尚未见报道。现将上海市儿童医院2014年诊断的1例Alagille综合征合并肝细胞癌的病例报告如下,并结合相关文献进行复习。
1 临床资料
患儿,女,6岁,因反复皮肤黄染6年余入院。患儿系G1P1,足月剖宫产(羊水少),出生体质量2 350 g,Apgar评分10分。生后3~5天发现患儿皮肤巩膜黄染,并呈进行性加重,期间伴有大便颜色变浅及尿色加深,一直于当地诊所予以中药治疗(具体不详),患儿黄疸反复并逐渐出现瘙痒,伴有身高、体质量增加缓慢。当地医院心脏超声提示:房间隔缺损、室间隔缺损、动脉导管未闭、主动脉弓及降主动脉稍细、右肺动脉狭窄。3岁时于当地医院行房间隔缺损、室间隔缺损修补术,动脉导管未闭纠治术。住院期间查肝功能提示总胆红素 232 μmol/L,直接胆红素 199.2 μmol/L,丙氨酸氨基转移酶 85 U/L,天冬氨酸氨基转移酶76 U/L,予以熊去氧胆酸口服,黄疸稍有消退,后家属坚持予以中药治疗(具体不详),效果不佳,期间间歇血胆红素升高,以直接胆红素为主,遂来我院。患儿父母体健,否认近亲结婚及家族性遗传病史。入院查体:体温37.4℃,身高 93 cm,体质量 13.8 kg,身高、体质量均低于同年龄、同性别儿童2个标准差;面容特殊,前额突出、眼眶深陷、尖下颌、鼻前端肥大;颜面部可见散在红色斑丘疹,伴有明显瘙痒,皮肤巩膜黄染++++,未及明显浅表淋巴结,胸前正中一陈旧性手术疤痕;双肺无殊,心前区可闻及Ⅱ、Ⅲ级收缩期杂音,肺动脉瓣区可闻及Ⅲ、Ⅳ级收缩期杂音;腹膨隆,肝肋下6 cm,质硬,剑突下6 cm,脾肋下3 cm,质软,余无殊。实验室检查:白细胞12.92×109/L,血红蛋白136 g/L,血小板655×109/L;总胆红素 135 μmol/L,直接胆红素 75.4 μmol/L,丙氨酸氨基转移酶37 U/L,天冬氨酸氨基转移酶156 U/L,γ-谷氨酰基转肽酶 196 U/L,总胆汁酸 360 μmol/L;凝血常规未见明显异常;甲胎蛋白 1 500.34 ng/mL(正常值<9 ng/mL)。腹部B超示肝内多发实性占位(图1),胰、脾、肾未见明显异常。腹部MRI示肝脏多发占位(图2)。心超提示修补术后未见残余分流,左心室收缩功能正常。眼底检查未见角膜后胚胎环。

图1 肝脏B超

图2 腹部MRI
经家属知情同意后,抽取患儿外周血4mL,EDTA抗凝,Sanger法测序Jagged 1基因。用PCR方法扩增出Jagged 1基因氨基酸编码区的26个外显子并测序,发现Jagged 1基因 c.1205delC,杂合型(图3)。结合Jagged 1基因检测结果和3个主要临床表现,即肝内胆汁淤积、心脏异常、特殊面容,患儿的Alagille综合征诊断成立。由于患儿影像学检查提示存在多发性肝细胞结节性病变,AFP明显升高,高度怀疑合并肝细胞癌。但因拟行肝移植术,未行肝穿刺术。最后,患儿因肝脏广泛占位,已无肝移植手术指征,于确诊半年后死亡。
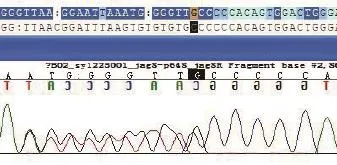
图3 患儿Jagged 1基因测序图
2 讨论
Alagille综合征为累及多系统的常染色显性遗传病,由人类Jagged 1或Notch2基因突变所致。75%的患儿可以存活至成年[3]。该基因编码Notch信号通路的一个配体,该信号通路对心脏、肝脏、骨骼、眼睛和面部等组织器官的生长发育起重要的调节作用。Jagged 1 是Notch受体的功能性配体,受体与配体相互作用启动下游信号转录,从而影响细胞的增殖、分化与凋亡。在体外,Notch信号控制细胞增殖和血管内皮细胞的迁移和分化;在体内,Notch 信号通路促进心脏中上皮-间质细胞转型,诱导血管的生成,并且Notch 信号可通过促进心肌再生、保护缺血心肌和抑制心脏成纤维细胞-肌成纤维细胞转化来修复心肌损伤[4]。目前研究发现,Notch信号在肝内胆管的生成及维持中起重要作用。Notch信号缺乏导致肝内胆管生成异常,胆管内皮细胞减少,并导致肝内胆管的主分支及中间支生成异常[5]。
肝细胞癌在儿童Alagille综合征中是罕见并发症,只有少数文献报道在Alagille综合征患儿中出现肝脏肿瘤(肝细胞癌和肝结节增生)。目前文献资料显示,17岁以下患儿仅有13例,13例患儿均有胆汁淤积和特殊面容,所有确诊肝细胞癌病例均得到组织学病理证实,仅1例有基因报告证实为Jagged 1基因突变;13例中最小为17个月的男性患儿,肝移植术后存活,肝脏病理提示为单个结节的肝细胞癌;11例患儿随访均死亡,1例未完全记录随访结果[6-13]。Alagille综合征并发肝细胞癌可能与大多数Alagille综合征有潜在肝硬化风险,肝硬化可能诱发恶性肿瘤有关。最近有证据显示Notch信号通路在肿瘤转化中的作用,能激活下游的Ras致癌基因。且在宫颈肿瘤、肾细胞癌、急性髓性白血病中已有报道[12]。
最新研究发现,确诊Jagged 1基因突变的患儿中,仅30%~50%的患者为遗传性突变,而50%~70%的患者为新生突变。迄今为止,已发现超过240种导致Alagille综合征的Jagged 1基因突变,以错义/无义突变占多数,其次为缺失/插入和剪接位点突变。Jagged 1基因中的某些突变,如3052 delGT、2504 del 5、1618insC、R184H、2871+1G>T等发生频率较高[14]。本例为c.1205delC缺失突变,此基因突变已有文献报道[15]。本例患儿依据临床表型,临床诊断考虑Alagille综合征后为进一步明确基因型,进行靶向基因测序,发现致病基因。高美玲等[16]对于行目标基因捕获测序发现突变的4例Alagille综合征患儿DNA采用Sanger法进行验证,结果发现与第二代测序也一致。由此进一步可以说明Alagille综合征患儿并非一定均使用二代测序,应该以临床表型为切入点,首先考虑靶向测序,只有临床表型复杂重叠,才考虑二代/靶向结合测序。
Alagille综合征目前以支持治疗为主,补充脂溶性维生素A、D、E、K和熊去氧胆酸利胆,当出现肝功能失代偿或瘙痒严重影响生长发育时可进行肝移植。合并肝细胞癌后,手术切除是首选,但是肝脏若出现多发性病灶或转移时则失去手术和肝移植机会。本例患儿出生后即出现黄疸,伴持续胆汁淤积伴瘙痒、生长发育落后并先天心脏异常,因确诊年龄较晚,未予以规范治疗和随访。
[1]Danks DM, Campbell PE, Jack I, et al. Studies of the aetiology of neonatal hepatitis and biliary atresia [J]. Arch Dis Child, 1977, 52(5): 360-367.
[2]Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and Management [J]. Eur J Hum Genet, 2012, 20(3): 251-257.
[3]Emerick KM, Rand EB, Goldmuntz E, et al. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis [J]. Hepatology, 1999, 29(3): 822-829.
[4]Zhou XL, Liu JC. Role of Notch signaling in the mammalian heart [J]. Braz J Med Biol Res, 2014, 47(1): 1-10.
[5]马艳立,宋元宗. Alagille 综合征诊断治疗进展[J].中国当代儿科杂志, 2014, 16 (11): 1188-1192.
[6]Kim B, Park SH, Yanq HR, et al. Hepatocellucar carcinoma occurring in alagille syndrome [J]. Pathol Res Pract, 2005, 201(1): 55-60.
[7]Kaufman SS, Wood RP, Shaw BW Jr, et al. Hepatocarcinoma in a child with the Alagille syndrome [J]. Am J Dis Child, 1987, 141(6): 698-700.
[8]Békássy AN1, Garwicz S, Wiebe T, et al. Hepatocellular carcinoma associated with arteriohepatic dysplasia in a 4-year old girl [J]. Med Pediatr Oncol, 1992, 20(1): 78-83.
[9]Rabinovitz M, Imperial JC, Schade RR, et al. Hepatocellular carcinoma in Alagille’s syndrome: a family study [J]. J Pediatr Gastroenterol Nutr, 1989, 8(1): 26-30.
[10]Chiaretti A, Zampino G, Botto L, et al. Alagille syndrome and hepatocarcinoma: a case report [J]. Acta Pediatr, 1992, 81(11): 937.
[11]Castaneda C, Fragoso T, Gra B, et al. Alagille’s syndrome in Cuba. A report of 9 cases [J]. Genetics, 1992, 46(4):341-346.
[12]Bhadri VA, Stormon MO, Arbuckle S, et al. Hepatocellular carcinoma in children with Alagille syndrome [J]. J Pediatr Gastroenterol Nutr, 2005,41(5):676-678.
[13]Wetli SC, Gralla ES, Schibli S, et al. Hepatocellular carcinoma and regenerating nodule in a 3-year-old child with Alagille syndrome [J]. Pediatr Radiol, 2010, 40910): 1696-1698.
[14]高辉,祁鸣. Alagille 综合征分子遗传学研究进展 [J].国际遗传学杂志, 2008, 31(5): 400-403.
[15]Lin HC, Le Hoang p, Hutchinson A, et al. Alagille syndrome in a Vietnamese cohort: mutation analysis and assessment of facial features [J]. Am J Med Genet A, 2012, 158A (5): 1005-1013.
[16]高美玲,钟雪梅,马昕,等. 目标基因捕获结合第二代测序技术诊断Alagille 综合征患儿四例[J].中华儿科杂志, 2016, 54(6): 441-444.
Alagille syndrome in a child combined with hepatocellular carcinoma: a case report and literature review
HU Hui1, YANG Yongchen2, ZHANG Hong2, ZHANG Ting1, XIAO Yongmei1
(1. Department of Gastroenterology, Hepatology, and Nutrition; 2. Department of Clinical Labortatory, Shanghai Children's Hospital Affiliated to Shanghai Jiao Tong University, Shanghai 200062, China)
ObjectiveTo explore the clinical manifestation, imaging examination, treatment and prognosis of Alagille syndrome in a child combined with hepatocellular carcinoma.MethodThe clinical manifestation, assistant examination and diagnosis of Alagille syndrome combined with hepatocellular carcinoma were analyzed in the child, and the pertinent literature were reviewed.ResultsThe 6-year-old girl was admitted to hospital for repeated jaundice, and had a history of cardiac surgery. After admission, the patient was found to have a typical face look such as frontal bossing, sunken eyes, pointed chin and hypertrophy of nasal tip. Blood biochemistry showed intrahepatic cholestasis and increased alpha-fetoprotein. Abdominal ultrasonography revealed diffuse multiple solid lesions in the liver. And magnetic resonance imaging of the liver indicated that the liver was enlarged and multiple solid space occupying masses. Jagged 1 gene detection showed heterozygosis mutation of c.1205delC.ConclusionAlagllie syndrome complicated with hepatocellular carcinoma in childhood is extremely rare, and early diagnosis and long-term follow-up are of positive signi fi cance for its treatment and prognosis.
Alagllie syndrome; hepatocellular carcinoma; child
10.3969/j.issn.1000-3606.2017.04.003
2016-09-08)
(本文编辑: 梁 华)
肖咏梅 电子信箱: xiaoym@shchildren.com.cn
猜你喜欢
中国现代医生(2022年19期)2022-11-04
中国典型病例大全(2022年7期)2022-04-22
中国生殖健康(2020年2期)2021-01-18
肝博士(2020年5期)2021-01-18
肝博士(2020年5期)2021-01-18
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
中国卫生标准管理(2015年16期)2016-01-20
西部中医药(2015年9期)2015-02-02
中国中医药现代远程教育(2014年20期)2014-03-01
