
Pdr1蛋白N端携带Flag标签的光滑假丝酵母的构建
2017-05-02 03:49:30田园倪琪彭奕冰
微生物与感染 2017年2期
田园,倪琪,彭奕冰
上海交通大学医学院附属瑞金医院检验科,上海 200025
·论著·
Pdr1蛋白N端携带Flag标签的光滑假丝酵母的构建
田园,倪琪,彭奕冰
上海交通大学医学院附属瑞金医院检验科,上海 200025
近年来,光滑假丝酵母已成为第二位引起侵袭性真菌感染的病原体。光滑假丝酵母对唑类药物(临床一线抗真菌药物)的敏感性低且易发生耐药,一直是研究的热点。介导光滑假丝酵母对唑类药物耐药的关键基因是转录因子pdr1,其功能性突变会使Pdr1蛋白功能过度活跃,导致下游唑类药物外排泵基因高表达,从而对唑类药物耐药。本研究利用同源重组技术,构建在基因pdr1的5′端定点插入3×Flag标签的重组菌株2a2和2b2,为后续利用免疫染色质共沉淀技术寻找Pdr1直接调控基因奠定基础。结果表明,3×Flag标签添加到Pdr1蛋白N端可成功表达Flag-Pdr1蛋白;与野生型菌株相比,表达Flag-Pdr1的菌株对氟康唑的耐药性增强。此外,与野生型菌株相比,表达Flag-Pdr1的菌株中cdr1和pup1基因表达水平显著上升,提示在Pdr1蛋白N端加Flag标签能使其功能活跃,表明N端对Pdr1蛋白功能具有重要意义。
光滑假丝酵母;pdr1基因;标签;基因敲除;耐药性
流行病学研究显示,近年来光滑假丝酵母(Candidaglabrata,C.glabrata)已成为第二大引起侵袭性真菌感染的病原体,其血流感染的病死率接近50%[1-3]。光滑假丝酵母感染流行的重要原因是其易对唑类药物耐药,而唑类药物是临床主要抗真菌药物。既往研究表明,光滑假丝酵母的唑类药物获得性耐药机制主要有两种[4-5],一种是光滑假丝酵母线粒体DNA丢失导致转录因子Pdr1过度活跃,在过度活跃的Pdr1蛋白作用下,其下游一些介导唑类药物耐药的基因(如ATP结合盒转运蛋白CDR1)大量转录[6],加速唑类药物排除,致使突变菌株对唑类药物的敏感性大大减低;另一种是pdr1基因单个碱基突变引起Pdr1蛋白功能过度活跃,这种使Pdr1蛋白功能过度活跃的突变称为Pdr1的功能性(gain of function,GOF)突变。
目前研究多局限于Pdr1 GOF突变引起的其他基因转录变化[7-9],罕有研究阐述作为转录因子的Pdr1直接结合的靶基因,而Pdr1蛋白单克隆抗体(简称单抗)缺乏是使这方面研究陷入困顿的主要原因。本研究利用两个可循环使用的筛选标记:ura3基因和诺尔斯菌素抗性基因nat1,进行2次同源重组和4次化学转化,在临床分离株背景下构建Pdr1蛋白N端带有Flag标签的菌株,蛋白免疫印迹法证实Flag-Pdr1蛋白能正确表达,表达Flag-Pdr1的菌株耐药性增强,且cdr1和pup1基因表达增加,提示Pdr1的N端修饰能使蛋白功能活跃。本研究以临床分离菌株为背景,利用可循环的筛选标记,结合基因敲除和同源重组技术,使Pdr1蛋白带有Flag标签,建立了光滑假丝酵母表达融合蛋白的方法体系,为进一步研究Pdr1蛋白功能打下了基础。
1 材料与方法
1.1 材料
光滑假丝酵母临床菌株2a和2b均由本实验室收集,其中2a为氟康唑敏感菌株,pdr1基因为野生型;2b为氟康唑耐药菌株,pdr1基因带有GOF突变。2a和2b分离自同一患者接受氟康唑治疗前后。质粒pBC1、pBC2和pRD16均由Irene Castano Navarro实验室馈赠。光滑假丝酵母标准菌株ATCC 2001为本实验室保存。质粒pBC1带有NAT1筛选标记,质粒pBC2带有NAT1标记和Flag标签。
1.2 方法
1.2.1 培养基 按文献配制酵母膏胨葡萄糖(yeast peptone dextrose,YPD)培养液及平板、含诺尔斯菌素YPD平板、LB培养液及平板、含氨苄西林LB培养液及平板、SD-URA平板、5-FOA平板[10-12]。
1.2.2 引物及测序 引物均由Primer3网站(http://primer3.ut.ee)设计,在美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI) BLAST网站(http://blast.ncbi.nlm.nih.gov/Blast.cgi)检查引物特异性后,由金唯智公司合成(表1)。本实验涉及的所有聚合酶链反应(polymerase chain reaction,PCR)产物和质粒均由铂尚生物技术(上海)有限公司测序。
1.2.3 菌株DNA抽提 菌株在YPD液体培养基中培养过夜,用1.5 mL EP管收集菌体,1 mL ddH2O重悬,再次离心,弃上清液;在EP管中加入200 μL溶胞液〔1 u/μL溶壁酶(lyticase)、1%十二烷基硫酸钠(sodium dodecyl sulfate,SDS)〕、200 μL苯酚/氯仿/异戊醇混合液(25∶24∶1)及200 μL 0.5 mm无菌玻璃珠(Sigma公司),水平振荡仪振荡5 min裂解细胞;氯仿抽提1次,无水乙醇沉淀,再用75%乙醇洗1次,ddH2O溶解,-20 ℃保存[13]。
1.2.4 质粒抽提和转化 从平板上挑取单克隆后接种于含有氨苄西林的LB液体培养基中过夜培养,按高纯度质粒抽提试剂盒说明书(天根生化科技有限公司)抽提质粒。感受态大肠埃希菌DH5α购自北京全式金生物技术有限公司,与转化连接产物或质粒混匀,置于冰上30 min,42 ℃热激90 s,冰上放置2 min,加入1 mL LB培养液复苏1 h,离心取100 μL沉淀,涂布于含有氨苄西林的LB平板。
1.2.5 光滑假丝酵母ura3基因敲除
1.2.5.1ura3基因上下游同源臂和筛选标记扩增ura3基因在光滑假丝酵母中编码乳清苷5′-磷酸脱羧酶。若ura3基因失活,光滑假丝酵母无法在缺乏尿苷或尿嘧啶的培养基中生长。因此,ura3基因敲除株只能在SD-URA平板(添加尿嘧啶的培养基)上生长。参照Schwarzmüller等的方法,利用融合PCR敲除ura3基因[14]。以ATCC 2001基因组DNA为模板,U1和U2为引物,扩增ura3起始密码子上游497 bp(UP);U5和U6为引物,扩增ura3终止密码子下游约303 bp(DOWN)。以pBC1为模板,U3和U4为引物,扩增筛选标记NAT片段(图1A),其中NAT筛选标记内nat1基因两端各有一个拷贝FRT。U2的5′端序列与U3的5′端序列反向互补,U4的5′端序列与U5的5′端序列反向互补。因此,扩增片段UP的3′端与筛选标记NAT片段5′端有39个bp互补序列,扩增片段DOWN的5′端与筛选标记NAT片段3′端有43个bp互补序列。PCR产物经1.5%琼脂糖凝胶电泳验证大小,并用QIAquick PCR Purification Kit(Qiagen)纯化。
1.2.5.2 融合PCR构建ura3基因敲除元件 将纯化后的UP、DOWN和NAT片段混合作为模板,用引物U1和U6进行融合PCR扩增(图1A)。PCR反应体系(50 μL):10×ExTaqBuffer 5 μL、2.5 mmol/L dNTP 4 μL、 10 μmol/L U1和U6各1 μL、UP和DOWN各50 ng、NAT片段170 ng,补 ddH2O至 50 μL。PCR反应条件: 95 ℃ 1 min预变性;95 ℃ 30 s,50 ℃ 30 s,72 ℃ 3 min,共30个循环;72 ℃ 7 min延伸。PCR产物经1.5%琼脂糖凝胶电泳确认为单一条带后用乙醇沉淀回收。
表1 引物序列
Tab.1 Primer sequences

PrimerSequenceU1ACCATTCCCGAGTTAAAACAATU2ACTGGCCGTCGTTTTACCTTGAGCTGGAGTATCCACTAAU3TTAGTGGATACTCCAGCTCAAGGTAAAACGACGGCCAGTU4TGTTGCTAGGTATGATCTAGCTTCGGAAACAGCTATGACCATGU5CATGGTCATAGCTGTTTCCGAAGCTAGATCATACCTAGCAACAU6CTTCTGATTAGAAGTCAGACGCURA3verFATCTTGCTGCCAGAAGGTCAURA3verRCCCCTCGAGGACGAAGTTCCTURAseqFCGAGAACCAATTGCATCATCCCURAseqRTCGGTTGTAAGATGATGTTGCTP1TCCCCGCGGCACCTGAGTTGTCCCGATGAP2CGCGGATCCGCATATTCTCTCAATAACGTAGCTP3CCGCTCGAGCAAACATTAGAAACTACATCAAAATCAAATCCAGGGGP4CGGGGTACCTCTCGCTGTCACTCCCTTTCAAmut1CACCTGAGTTGTCCCGATGAmut2GAACTTCGGATCATATAACTTGTCATCGTCATCTTTGTAGTCAGCmut3GACAAGTTATATGATCCGAAGTTCCTATACTTTCTAGAGAATAGGAmut4TCTCGCTGTCACTCCCTTTCAAM13FWGTAAAACGACGGCCAGTGM13RVGGAAACAGCTATGACCATGNtag5’verFTGTAAAACGACGGCCAGTGANtag5’verRTGGTTCAAAAGCGCACGAAGPdr1seqFACAAGCATAGAGGCGCTGTTPdr1seqRGGAACCTGGGACCAACATGA
表2 菌株
Tab.2 Strains used in this study

菌株表型亲代菌株用途来源2a———临床菌株2b———临床菌株2a1ura3Δ∷FRT∶NAT∶FRT2a带nat1基因的ura3基因敲除株本研究2b1ura3Δ∷FRT∶NAT∶FRT2b带nat1基因的ura3基因敲除株本研究2a2ura3Δ∷FRT2a1ura3基因敲除株本研究2b2ura3Δ∷FRT2b1ura3基因敲除株本研究2a3ura3Δ∷FRT∶3×Flag-Pdr12a2Pdr1蛋白N端表达3×Flag的菌株本研究2b3ura3Δ∷FRT∶3×Flag-Pdr12b2Pdr1蛋白N端表达3×Flag的菌株本研究

A: Construction ofura3 gene deletion with fusion PCR. B: Homologous recombination ofura3 gene deletion cassette.
图1ura3基因敲除元件的构建和ura3基因敲除
Fig.1 Methods of construction ofura3 deletion cassette andura3 gene deletion
1.2.5.3 光滑念珠菌ura3基因敲除株的构建 按Ueno等的醋酸锂转化方法[15],将ura3基因敲除元件转入2a和2b(图1B),用无菌玻璃珠在YPD-NAT平板上均匀涂布菌液,30 ℃培养48 h;挑取单克隆,在YPD中增菌后抽提基因组,在接头处设计引物URA3 verF和URA3 verR,确认敲除元件同源重组成功(图2A),获得ura3基因敲除的2a和2b,命名为2a1和2b1。所有构建的菌株在表2中列出。
1.2.5.4 NAT筛选标记的循环利用 参照Navarro的方法。由于pRD16带有酿酒酵母Flp1重组酶,该酶能识别nat基因两端的34 bp FRT,催化重组,使两个FRT之间的nat1基因及一个FRT拷贝丢失,留下一个FRT标记。此外,pRD16带有URA3标记。因此,ura3缺陷菌株若被转化入pRD16,能在尿嘧啶缺乏的条件下生存(图2B)。同样,用醋酸锂转化方法将质粒pRD16转化入2a1和2b1,在SD-URA平板上用玻璃珠涂布菌液,30 ℃培养48 h,挑取单克隆,用URA seqF和URA seqR验证(图2C)。将验证的nat丢失菌株转接到5-FOA平板,使pRD16质粒丢失,获得不带NAT筛选标记的ura3基因敲除株2a2和2b2。
1.2.6 构建表达Pdr1 N端带有Flag标签的光滑假丝酵母
1.2.6.1 构建Pdr1 N端加Flag元件 以2a基因组DNA为模板,P1和P2为引物,扩增pdr1基因起始密码子前约500 bp片段(pdr1 UP);P3和P4为引物,扩增pdr1基因起始密码子后约500 bp片段(pdr1 DOWN)。引物P1和P2的5′端带有SacⅡ和BamHⅠ酶切位点和保护碱基,引物P3和P3的5′端带有KpnⅠ和XhoⅠ酶切位点和保护碱基。因此,pdr1 UP片段5′ 端和3′端分别带有SacⅡ和BamHⅠ酶切位点,pdr1 DOWN片段5′端和3′端带有KpnⅠ和XhoⅠ酶切位点。将pdr1 UP片段和pBC2质粒用SacⅡ和BamHⅠ双酶切过夜,所有酶切产物经1.5%琼脂糖凝胶电泳,在紫外灯下用刀片切下目的条带,QIAquick Gel Extraction Kit(Qiagen)纯化,DNA Ligation Kit Ver.2.1(TaKaRa)将pdr1 UP酶切回收产物与pBC2酶切回收产物连接,并转化感受态DH5α。挑取单克隆转化子,抽取质粒DNA,用引物M13 FW和M13 RV进行PCR扩增验证,获得质粒pBC21(图3)。将质粒pBC21和pdr1 DOWN片段用KpnⅠ和XhoⅠ双酶切切开。同样方法,将pdr1 DOWN片段连入pBC21质粒,用M13 FW和M13 RV引物PCR扩增验证,获得质粒pBC22。由于pBC2质粒中Flag序列后有两个终止密码子,用mut1、mut2引物对扩增mutUP,mut3、mut4引物对扩增mutDOWN。mutUP的3′端与mutDOWN的5′端有40 bp反向互补序列,通过在mut2和mut3引物对上设计突变位点,将Flag序列后的终止密码子突变,再利用融合PCR技术以mutUP和mutDOWN为模板,mut1和mut4为引物,扩增Pdr1 N端加Flag元件(图3B)。
1.2.6.2 表达Pdr1 N端带有Flag标签的光滑假丝酵母的构建 用醋酸锂方法使N端加Flag元件转化菌株2a2和2b2,并在YPD-NAT平板上进行筛选,转化子用Ntag5’verF和Ntag5’verR引物对PCR扩增验证(图4)。将pRD16转化入正确的转化子,在SD-URA平板上进行筛选并挑取单克隆,用Pdr1 seqF和Pdr1 seqR引物对验证正确克隆后,在5-FOA平板上转接1次,使pRD16质粒丢失,获得不带NAT筛选标记、ura3基因敲除、pdr1基因N端带有Flag序列的菌株2a3和2b3。
1.2.6.3 Pdr1蛋白N端Flag表达的验证 菌株2a3、2b3、2a2和2b2在YPD中培养过夜,0.1×OD600时转接,长至1×OD600后收菌;用冰水洗1次,1 mL冰水重悬,并加入2 mol/L NaOH 150 μL,冰上放置30 min,期间振荡2~3次;再加入150 μL 50%三氯乙酸,冰上放置30 min,期间振荡2~3次;最大转速离心取沉淀,加入适量的HU Buffer,于65 ℃金属浴加热溶解,再次离心去上清液, -80 ℃保存。将2a3和2b3的蛋白经SDS-聚丙烯酰胺凝胶电泳(SDS-polyacrylamide gel electrophoresis,SDS-PAGE),转移至聚偏氟乙烯(polyvinylidene fluoride,PVDF)膜,用1∶10 000稀释的Flag M2单抗为一抗(Sigma公司),Tubulin(1∶2 000稀释,Novus公司)为内参,辣根过氧化物酶(horseradish peroxidase,HRP)标记山羊抗小鼠(与Flag M2一抗对应)为二抗,HRP标记山羊抗大鼠(与Tubulin一抗对应)为二抗,经增强化学发光(enhanced chemiluminescence,ECL)显色,检测Flag-Pdr1的表达[16]。

A: Verification of correct integration ofura3 deletion cassette. B: Flipping of NAT marker. C: Verification of flipping NAT marker with PCR.
图2ura3 基因敲除株的验证和NAT筛选标记的循环利用
Fig.2 Verification ofura3 knock-out transformants and recycling of NAT marker
1.2.7 构建菌株表型验证
1.2.7.1 滴板实验验证 菌株过夜培养于YPD中,离心收集后用灭菌水重悬,调整菌液密度至2×107/mL,用水依次10倍稀释成4个密度,每个密度取5 μL点于含氟康唑的培养板上,30 ℃培养48 h后拍摄菌落形态[17]。
1.2.7.2 荧光定量PCR 菌株于YPD中培养至对数生长期,离心收集,用Yeast RNAiso Kit(TaKaRa公司)提取mRNA,PrimeScriptTMRT reagent Kit with gDNA Eraser(TaKaRa公司)将mRNA反转成cDNA,用SYBR®Premix Ex TaqTMII(Tli RNaseH Plus)于Roche LightCycler 480系统定量检测cDNA。
2 结果
2.1 2a和2b菌株的鉴定
经鉴定,2a和2b能在CHROMagar假丝酵母显色培养基、尿嘧啶营养缺陷SD-URA平板上生长,但无法在YPD-NAT平板及5-FOA平板上生长。
2.2ura3基因敲除元件的鉴定
ura3基因敲除元件UP-nat-DOWN由上段ura3 UP(497 bp)、nat(1 764 bp)及下段ura3 DOWN(303 bp)3段经融合PCR构成,全长2 564 bp。如图5A所示,电泳条带大小正确。

A: Construction of plasmid pBC22. B: Mutation of stop codons with fusion PCR. C: Verification of plasmid pBC21 and pBC22.
图3 Pdr1蛋白N端加Flag元件的构建原理
Fig.3 Method of construction of N-terminally Flag-tagged cassette of Pdr1
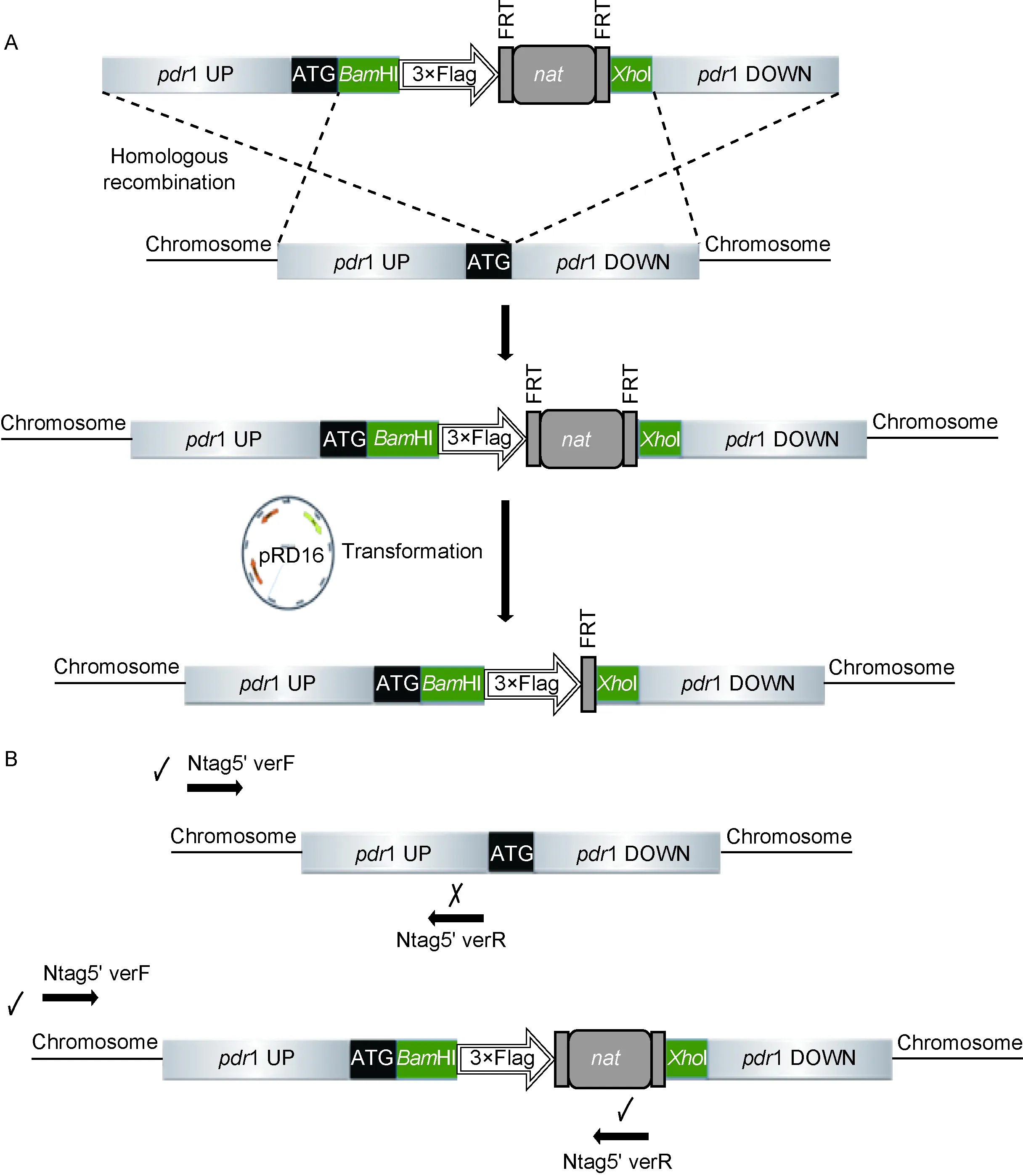
图4 Pdr1蛋白N端加Flag元件转化及转化子验证原理
Fig.4 Methods of transforming N-terminally Flag-tagged cassette of Pdr1 and verification of correct integration
2.3ura3基因敲除株的鉴定
验证ura3基因敲除株时,用接头处引物对URA3 verF和URA3 verR 进行PCR扩增, PCR产物大小应为860 bp,电泳结果(图5B)与预期大小一致,产物经测序与预期一致,ura3基因开放读码框成功被NAT筛选标记替代。ura3基因敲除株2a1和2b1无法在营养缺陷培养基SD-URA平板上生长,但可在5-FOA平板上生长。
2.4ura3基因敲除株NAT筛选标记的剔除
pRD16转入ura3基因敲除菌株后,在SD-URA平板上挑选单克隆并抽提基因组,用引物对URA3 seqF和URA3 seqR验证,PCR产物应为304 bp,电泳结果与预期一致(图5B),NAT筛选标记成功切除。在5-FOA平板上转接NAT筛选标记切除株,使pRD16质粒丢失。
2.5pdr1基因N端加Flag元件的鉴定
以质粒pBC2为模板,M13 FW和M13 RV为引物,进行PCR扩增,产物长度为1 868 bp,而pdr1 UP片段长度为590 bp,因此pdr1 UP片段连入pBC2质粒构建为pBC21后,M13 FW和M13 RV扩增产物应为2 458 bp;pdr1 DOWN片段长度为577 bp,其连入pBC21质粒构建为pBC22质粒后,M13 FW和M13 RV扩增产物应为3 035 bp。电泳结果均符合预期(图6A)。融合PCR介导的终止密码子突变中,mutUP长度为712 bp,mutDOWN长度为2 197 bp,两端融合后大小为2 869 bp,电泳结果符合预期(图6B)。
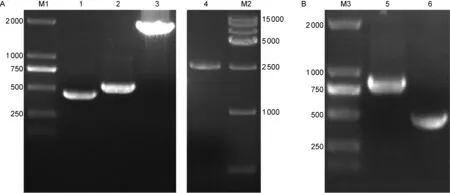
A:ura3 deletion cassette was constructed with fusion PCR. Size of UP was 497 bp (lane 1), DOWN 303 bp (lane 2) andnat1 764 bp (lane 3). Fusion PCR was done with UP, DOWN andnatas templates, and size of PCR product should be as long as 2 564 bp. B: Verification ofura3 knock-out strain and flipping away NAT marker. Correct integration of deletion cassette was verified by PCR with URA3 verF and URA3 verR, and PCR product size should be as long as 860 bp. NAT marker was flipped away by transforming correct transformants with pRD16, and cells were selected on SD-URA plates. URA3 seqF and URA3 seqR were used to amplify the genomic DNA of transformants, and expected PCR product size was as long as 304 bp.
图5ura3基因敲除元件构建及敲除株的鉴定
Fig.5 Identification ofura3 deletion cassette andura3 knock-out mutants

A: Construction of plasmids pBC21 and pBC22. Plasmids pBC21 and pBC22 were constructed and verified by PCR with primers M13 FW and M13 RV. PCR products were separated by electrophoresis. PCR product should be as long as 1 868 bp (lane 3) if pBC2 was used as template, 2 458 bp (lane 2) if pBC21 was used as template, and 3 035 bp (lane 1) if PBC22 was used as template. B: PCR mediated point mutation. Point mutation to construct Pdr1 N-terminally Flag-tagged cassette was done with PCR. Size ofmutUP was 712 bp (lane 4), and formutDOWN 2 179 bp (lane 5). Fusion PCR withmutUP andmutDOWN as template produced Pdr1 N-terminally Flag-tagged cassette, which was as long as 2 869 bp (lane 6 ). C: Verification of correct transformants with N-terminally Flag-tagged Pdr1. Correct integration of Pdr1 N-terminally Flag-tagged cassette was verified by PCR with Ntag5’verF and Ntag5’verR, and PCR product size was as long as 814 bp (lane 7). NAT marker was flipped away as described, and verification was done by PCR with Pdr1 seqF and Pdr1 seqR. The product size was as long as 673 bp.
图6 Pdr1蛋白N端加Flag元件构建及表达Flag-Pdr1菌株的鉴定
Fig.6 Construction of Pdr1 N-terminally Flag-tagged cassette
2.6 Pdr1蛋白N端表达Flag菌株的鉴定
用醋酸锂法转化2a2和2b2后,将YPD平板上挑选出的单克隆通过PCR检测正确的整合,Ntag5’verF和Ntag5’verR引物对的产物大小应为814 bp(图6C),电泳结果符合预期。用质粒pRD16去除NAT筛选标记后,在SD-URA平板上挑取单克隆,抽提基因组DNA,用引物对Pdr1 seqF和Pdr1 seqR扩增后,产物大小应为673 bp(图6C),电泳结果符合预期。
2.7 Pdr1蛋白N端表达Flag标签的验证
在对数生长期抽提2a3、2b3及2a菌株蛋白作为阴性对照,经10% SDS-PAGE电泳后,蛋白被转移到PVDF膜上,封闭,加入一抗、二抗后显影。结果显示,菌株2a3和2b3在蛋白Marker(120 000)处有条带,2a菌株蛋白没有任何条带,符合预期结果(图7),表明2a3和2b3菌株能表达N端带有Flag标签的Pdr1蛋白。

图7 蛋白免疫印迹法检测Flag-Pdr1蛋白的表达
Fig.7 Detection of protein expression of Flag-Pdr1 by Western blotting
2.8 菌株表型的验证
2.8.1 滴板实验验证Flag-Pdr1菌株表型 Pdr1蛋白在光滑假丝酵母中主要介导耐药表型。用滴板实验验证Pdr1蛋白N端加上Flag标签后对菌株表型的影响,验证2a、2b、2a2、2a3、2b2及2b3菌株的药物敏感性(氟康唑),结果如图8所示。2a与2a2在含氟康唑的YPD平板上生长没有差异,而2a3比2a和2a2对氟康唑更耐药,但耐药率低于2b。2b、2b2与2b3在含氟康唑的YPD平板上生长没有差异;而微量肉汤稀释法显示2b3的MIC80(氟康唑)值为256 μg/mL,2b2的MIC80(氟康唑)为128 μg/mL(表3)。
表3 菌株MIC80(氟康唑)
Tab.3 MIC80values of fluconazole

菌株来源FluconazoleMIC80(μg/mL)2a临床 82b临床 1282a1本研究82a2本研究82a3本研究322b1本研究1282b2本研究1282b3本研究256

YPD, YPD agar plates without fluconazole; FLC64, YPD plates with 64 μg/mL fluconazole.
图8ura3基因敲除株及表达Flag-Pdr1蛋白菌株的氟康唑药敏表型鉴定
Fig.8 Fluconazole-susceptibility ofura3 gene deleted strains and strains expressing Flag-Pdr1
2.8.2 实时定量PCR验证Flag-Pdr1菌株表型 与2a相比,2a3中cdr1和pup1基因的表达水平显著增加;与2b相比,2b3中cdr1和pup1基因的表达水平显著增加(P<0.05)(图9)。

For each sample, three biological replicates were included. Statistical significance was determined with Student’s unpairedttest.*P<0.05,**P<0.01. Error bars indicate the standard deviations.
图9 Flag-Pdr1菌株与野生型菌株基因表达水平的差异
Fig.9 Relative gene expression levels in strains expressing Flag-Pdr1 and wild-type
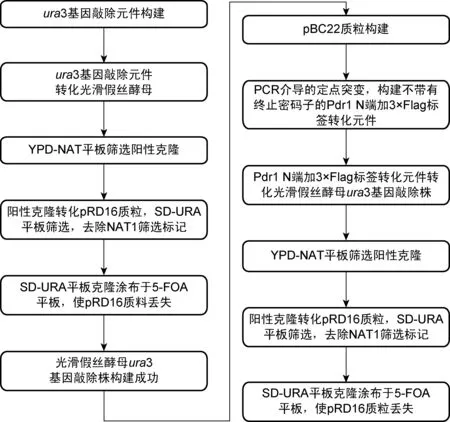
图10 在野生型光滑假丝酵母中构建Pdr1融合蛋白的流程图
Fig.10 Flow chart of tagging Pdr1 in clinic isolatedC.glabrata
3 讨论
光滑假丝酵母所致系统性感染的病死率达50%[1]。由于其天然对唑类药物不敏感,极易对唑类药物耐药,导致其感染治疗很困难。光滑假丝酵母的耐药机制主要是药物外排泵表达增多,其中cdr1基因编码的ABC转运蛋白表达增加在唑类药物耐药发生中极为重要。Ferrari等发现,调控cdr1基因表达的转录因子pdr1基因突变会导致cdr1表达持续增高。pdr1基因突变被称为GOF突变,可导致光滑假丝酵母对唑类药物尤其是氟康唑耐药。除耐药性改变外,GOF突变菌株的毒力和黏附力也增强[9]。这些研究表明,pdr1的GOF突变在光滑假丝酵母感染宿主过程中起重要作用。
本研究利用酵母高频同源重组的原理,将蛋白标签与目的蛋白融合表达。在白假丝酵母,同源重组最常用的筛选标记是SAT1-flipper组件,该组件同时带有nat1基因和flp1基因。当其整合至酵母后,菌株表现出诺尔斯菌素耐药表型;当转化子在麦芽糖培养基中培养时,Flp1表现出微弱的活性,将筛选标记去除。Flp1的活性在麦芽糖培养基中太过微弱,给筛选标记的循环使用带来很大困难[18],因此本研究参考Beese-Sims的基因敲除方法[19],通过4次转化光滑假丝酵母,使目的蛋白N端表达Flag标签。第1次转化以nat1作为筛选标记,敲除ura3基因;第2次转化以ura3基因作为筛选标记,将第1次转化在酵母基因组ura3基因座上的NAT标记用pRD16质粒上的Flp1重组酶剔除,使NAT筛选标记能循环使用;第3次转化再次以NAT作为筛选标记,将Flag标签核酸序列融合到pdr1基因N端;第4次转化再次用质粒pRD16上的Flp1重组酶剔除位于pdr1基因N端的NAT筛选标记,使Flag标签能在pdr1基因启动子作用下与Pdr1蛋白融合表达。与SAT1-flipper筛选标记相比,本研究需两种筛选标记,多次转化,较为繁琐。在SAT1-flipper筛选中,通过在麦芽糖培养基催化下启动Flp1活性,在低浓度诺尔斯菌素平板上筛选菌落较大的单克隆,会大大增加假阳性的可能;而本研究通过将质粒pRD16转入去除NAT筛选标记,由于pRD16的转化效率非常高,且flp1基因由强启动子启动,NAT筛选标记的剔除十分简便,其成功率几乎为100%,假阳性极低,重复性高,可靠性好(图10)。
滴板实验证实,与野生型菌株相比,表达Flag-Pdr1的菌株耐药性显著增加。实时定量PCR进一步证实,与野生型菌株相比,表达Flag-Pdr1的菌株中介导对唑类药物耐药的cdr1基因表达水平显著增加。结果表明,在Pdr1蛋白N端添加Flag标签不仅使唑类药物敏感菌变得耐药,还使唑类药物耐药菌变得更耐药,提示N端修饰Pdr1能使其功能更活跃,与pdr1基因GOF突变类似。后续可通过免疫染色质沉淀实验,用anti-Flag抗体免疫沉淀中与N端带有3×Flag标签Pdr1结合的DNA片段,验证N端带有Flag标签的Pdr1与下游基因启动子是否具有更强的结合力。
本研究在临床菌株背景下,利用两个筛选标记建立了一个可在同一株菌中同源重组多次的方法体系,构建了表达N端带有Flag标签的Pdr1蛋白的光滑假丝酵母。滴板实验和实时定量PCR证实N端带有Flag标签的Pdr1功能变得更活跃,可进一步对Pdr1蛋白N端进行功能研究。关于GOF突变后的Pdr1蛋白结合下游靶基因发生的改变,以及GOF突变后的Pdr1蛋白会与哪些新的蛋白相互作用,需进一步研究。
[1] Zaoutis TE, Argon J, Chu J, Berlin JA, Walsh TJ, Feudtner C. The epidemiology and attributable outcomes of candidemia in adults and children hospitalized in the United States: a propensity analysis [J]. Clin Infect Dis, 2005, 41(9): 1232-1239.
[2] Borah S, Shivarathri R, Srivastava VK, Ferrari S, Sanglard D, Kaur R. Pivotal role for a tail subunit of the RNA polymerase II mediator complex CgMed2 in azole tolerance and adherence in Candida glabrata [J]. Antimicrob Agents Chemother, 2014, 58(10): 5976-5986.
[3] Nishikawa JL, Boeszoermenyi A, Vale-Silva LA, Torelli R, Posteraro B, Sohn YJ, Ji F, Gelev V, Sanglard D, Sanguinetti M, Sadreyev RI, Mukherjee G, Bhyravabhotla J, Buhrlage SJ, Gray NS, Wagner G, Näär AM, Arthanari H. Inhibiting fungal multidrug resistance by disrupting an activator-mediator interaction [J]. Nature, 2016, 530(7591): 485-489.
[4] Vermitsky JP, Edlind TD. Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor [J]. Antimicrob Agents Chemother, 2004, 48(10): 3773-3781.
[5] Tsai HF, Krol AA, Sarti KE, Bennett JE. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants [J]. Antimicrob Agents Chemother, 2006, 50(4): 1384-1392.
[6] Sanglard D, Ischer F, Calabrese D, Majcherczyk PA, Bille J. The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents [J]. Antimicrob Agents Chemother, 1999, 43(11): 2753-2765.
[7] Caudle KE, Barker KS, Wiederhold NP, Xu L, Homayouni R, Rogers PD. Genomewide expression profile analysis of the Candida glabrata Pdr1 regulon [J]. Eukaryot Cell, 2011, 10(3): 373-383.
[8] Tsai HF, Sammons LR, Zhang X, Suffis SD, Su Q, Myers TG, Marr KA, Bennett JE. Microarray and molecular analyses of the azole resistance mechanism in Candida glabrata oropharyngeal isolates [J]. Antimicrob Agents Chemother, 2010, 54(8): 3308-3317.
[9] Ferrari S, Ischer F, Calabrese D, Posteraro B, Sanguinetti M, Fadda G, Rohde B, Bauser C, Bader O, Sanglard D. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence [J]. PLoS Pathog, 2009, 5(1): e1000268.
[10] Gutiérrez-Escobedo G, Orta-Zavalza E, Castao I, De Peas A. Role of glutathione in the oxidative stress response in the fungal pathogen Candida glabrata [J]. Curr Genet, 2013, 59(3): 91-106.
[11] Healey KR, Zhao Y, Perez WB, Lockhart SR, Sobel JD, Farmakiotis D, Kontoyiannis DP, Sanglard D, Taj-Aldeen SJ, Alexander BD, Jimenez-Ortigosa C, Shor E, Perlin DS. Prevalent mutator genotype identified in fungal pathogen Candida glabrata promotes multi-drug resistance [J]. Nat Commun, 2016, 7: 11128. doi: 10.1038/ncomms11128.
[12] Poláková S, Blume C, Zárate JA, Mentel M, Jørck-Ramberg D, Stenderup J, Piskur J. Formation of new chromosomes as a virulence mechanism in yeast Candida glabrata [J]. Proc Natl Acad Sci USA, 2009, 106(8): 2688-2693.
[13] van Burik JA, Schreckhise RW, White TC, Bowden RA, Myerson D. Comparison of six extraction techniques for isolation of DNA from filamentous fungi [J]. Med Mycol, 1998, 36(5): 299-303.
[14] Schwarzmüller T, Ma B, Hiller E, Istel F, Tscherner M, Brunke S, Ames L, Firon A, Green B, Cabral V, Marcet-Houben M, Jacobsen ID, Quintin J, Seider K, Frohner I, Glaser W, Jungwirth H, Bachellier-Bassi S, Chauvel M, Zeidler U, Ferrandon D, Gabaldón T, Hube B, d’Enfert C, Rupp S, Cormack B, Haynes K, Kuchler K. Systematic phenotyping of a large-scale Candida glabrata deletion collection reveals novel antifungal tolerance genes [J]. PLoS Pathog, 2014, 10(6): e1004211.
[15] Ueno K, Uno J, Nakayama H, Sasamoto K, Mikami Y, Chibana H. Development of a highly efficient gene targeting system induced by transient repression of YKU80 expression in Candida glabrata [J]. Eukaryot Cell, 2007, 6(7): 1239-1247.
[16] Lavoie H, Sellam A, Askew C, Nantel A, Whiteway M. A toolbox for epitope-tagging and genome-wide location analysis in Candida albicans [J]. BMC Genomics, 2008, 9:578. doi: 10.1186/1471-2164-9-578.
[17] Alonso-Monge R, Navarro-García F, Román E, Negredo AI, Eisman B, Nombela C, Pla J. The Hog1 mitogen-activated protein kinase is essential in the oxidative stress response and chlamydospore formation in Candida albicans [J]. Eukaryot Cell, 2003, 2(2): 351-361.
[18] Reuss O, Vik A, Kolter R, Morschhäuser J. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans [J]. Gene, 2004, 341: 119-127.
[19] Beese-Sims SE, Pan SJ, Lee J, Hwang-Wong E, Cormack BP, Levina DE. Mutants in the Candida glabrata glycerol channels are sensitized to cell wall stress [J]. Eukaryot Cell, 2012, 11(12): 1512-1519.
. PENG Yibing, E-mail: pyb9861@sina.com
Amino terminus Flag tagging on Pdr1 inCandidaglabrata
TIAN Yuan, NI Qi, PENG Yibing
ClinicalLaboratory,RuijinHospital,ShanghaiJiaoTongUniversitySchoolofMedicine,Shanghai200025,China
Candidaglabrata(C.glabrata) is emerging as the second most common cause for invasive candidiasis and early studies indicate that it is relatively insensitive to azole treatment. Transcription factor Pdr1 is the key regulator of azole resistance genes inC.glabrata. In this study, a Flag sequence was introduced into amino terminus ofpdr1 gene at its native locus in chromosome through homologous recombination in clinical isolates ofC.glabrata. Western blotting analysis with anti-Flag antibody showed a single immunoreactive species matching the predicted size of Pdr1. Spot assay showed that mutants expressing Flag-Pdr1 were more resistant to fluconazole than wild-type. Real-time quantitative polymerase chain reaction showed that gene expression levels ofcdr1 andpup1 in mutants expressing Flag-Pdr1 were significantly increased than those in wild-type strains.
Candidaglabrata;pdr1; Tag; Gene deletion; Resistance
国家自然科学基金 (81371873、81301462、81572053)
彭奕冰
2016-12-21)
猜你喜欢
昆明医科大学学报(2022年1期)2022-02-28 07:43:38
中国民间疗法(2021年8期)2021-07-22 05:53:28
数学大王·中高年级(2021年4期)2021-04-27 11:21:14
家庭影院技术(2019年8期)2019-08-27 02:44:52
中国调味品(2017年2期)2017-03-20 16:18:25
创新作文(小学版)(2016年16期)2016-11-11 05:47:54
现代检验医学杂志(2016年5期)2016-08-20 03:17:04
中国科技信息(2015年2期)2015-11-16 08:18:32
药学与临床研究(2015年4期)2015-06-05 11:35:51
计算机与网络(2014年9期)2014-03-25 10:57:13
