
重组琼胶酶rAgaN3基因的生物信息学分析
2017-04-10 06:10:45洪晓昆鄢仁祥
生物信息学 2017年1期
谢 勇,洪晓昆,鄢仁祥,林 娟
(福建省海洋酶工程重点实验室(福州大学),福州 350116)
重组琼胶酶rAgaN3基因的生物信息学分析
谢 勇,洪晓昆,鄢仁祥,林 娟
(福建省海洋酶工程重点实验室(福州大学),福州 350116)
利用生物信息学软件和数据库对从Microbulbifersp. BN中得到的琼胶酶rAgaN3全长基因进行预测分析,结果表明:重组琼胶酶rAgaN3理论分子量为31.243 kDa,理论等电为4.81,不稳定系数为26.23,脂肪系数为62.35,平均疏水性系数为-0.662,无跨膜结构域,无信号肽;二级结构表明该蛋白无螺旋结构,有15个折叠结构,其余均为卷曲结构;序列相似性分析表明,蛋白rAgaN3属于糖苷水解酶GH16家族,为β-琼胶酶;以同源蛋白3wz1A(同源性88%)为模板,通过同源建模构建出了蛋白三级结构,并用拉式图进行了结构检验。琼胶酶rAgaN3基因的生物信息学分析,为琼胶酶的异源表达提供了指导,为琼胶酶的定点突变、深入研究其结构和功能的关系打下良好基础。
琼胶酶;基因分析;生物信息学;蛋白结构
琼脂存在于石花菜科和龙须菜科红藻细胞壁中,主要由琼脂糖和琼脂胶组。其中琼脂糖是由(1-3)-O-β-D-半乳糖和(1-4)-O-3,6-内醚-α-L-半乳糖交替组成的线形链状分子[1],是目前世界上应用最广泛的海藻多糖之一。琼胶具有胶凝性和凝胶的稳定性,因而在食品工业中常被用作胶凝剂、增稠剂、乳化剂、增量剂、助悬剂、水分保持剂、稳定剂、赋形剂等。
琼胶酶是一种能够降解琼胶,产生琼胶寡糖的酶总称。根据琼胶酶作用糖苷键的不同可以分为两类:α-琼胶酶和β-琼胶酶[2-3]。目前已报道的琼胶酶大多来源于海洋微生物,其中α-琼胶酶主要来自假单胞菌属、单胞菌属和弧菌属,β-琼胶酶主要来源于弧菌属、交替单胞菌属[4]。在许多软体动物的消化液的微生物中可以分离得到琼胶酶,比如滨螺属(Littorinastriata)、海兔属(Aplysiadactylomela)、冠海詹属(Diademaantillarum)、鲍属(Haliotiscoccinea)[5-6]。琼胶酶可用做海藻降解的工具酶,多用于多糖结构的研究[7],琼胶寡糖的制备以及在分子生物学方面也多有应用[8]。
目前关于琼胶酶的研究主要集中在菌种选育和基因工程菌构建上,传统菌株选育出的菌株所产琼胶酶常存在酶活低、稳定性差等缺点,通过基因克隆表达重组琼胶酶蛋白已经成为趋势。从1987年Buttner等[9]首次实现琼胶酶基因的异源表达开始,已经有一定数量的琼胶酶基因被研究,但是国内对琼胶酶基因的研究还不多。课题组从福建漳江口红树林泥样中分离获得一株产琼胶酶菌株,并进行了形态学和16SrRNA鉴定,确定菌种为微球茎菌属(Microbulbifersp.),并通过TAIL-PCR方法克隆得到基因全长。目前报道的微球茎菌属重组琼胶酶还较少,且与此基因编码的蛋白质序列有高同源性的蛋白3wz1A的结构已经得到解析[10],为rAgaN3的后续研究提供了便利。本文利用生物信息学软件和数据库,预测其理化性质及结构信息,为琼胶酶基因的异源表达和琼胶酶结构与功能关系研究奠定基础。
1 材料与方法
1.1 实验材料
菌株:Microbulbifersp. BN从福建漳江口红树林泥样中分离获得,利用 TAIL-PCR方法从中克隆得到琼胶酶基因agaN3序列全长。
1.2 分析方法
(1)利用 NCBI的 blast(http://www.ncbi.nlm.nih.gov/)和 MEGA5.2构建系统进化树;
(2)利用 ProtParam[11](http://web.expasy.org/protparam/)分析蛋白质的理化性质,包括相对分子质量、氨基酸组成、等电点、不稳定系数、总平均亲水性等;
(3)利用ProtScale[11](http://web.expasy.org/protscale/)分析蛋白质疏水性;
(4)利用TMHMM[12](http://www.cbs.dtu.dk/services/TMHMM/)分析蛋白质跨膜区域;
(5)利用PSIPRED[12](http://bioinf.cs.ucl.ac.uk/psipred/)分子蛋白质二级结构,比如分螺旋、卷曲、无规则卷曲等;
(6)利用PredictProtein[13](https://www.predictprotein.org/)分析跨膜螺旋区域、二硫桥以及结合位点等性质;
(7)利用Signal P[14](http://www.cbs.dtu.dk/services/SignalP/)分析信号肽;
(8)利用NCBI(http://www.ncbi.nlm.nih.gov/cdd/)进行保守结构域搜索;
(9)利用SMART[15](http://smart.embl-heidelberg.de/)预测结构域;
(10)利用CAZY[16]数据库(http://www.cazy.org/GH16.html/)查询糖苷水解酶信息;
(11)利用TargetP 1.1[13](http://www.cbs.dtu.dk/services/TargetP/)分析亚细胞定位及导肽;
(12)利用 SWISS-MODEL[17](http://www.swissmodel.expasy.org/)进行同源建模;
(13)利用SAVES[18]服务器 (http://services.mbi.ucla.edu/SAVES/) 对蛋白结构残基角度检验;
(14)利用PROSA[19]程序(https://prosa.services.came.sbg.ac.at/prosa.php)对蛋白结构能量进行检验。
2 结果分析
2.1 基因序列分析
rAgaN3的 DNA序列共有 834 bp,其中第 831~834位为终止密码子TAA,该蛋白含有 277个氨基酸。基因及蛋白序列如图1。
从 NCBI的 BLASTp的序列比对结果中选择12条Identity比较高的序列,与rAgaN3的序列在Clustal X 上进行序列比对,然后利用MEGA5.2构建系统进化树。系统进化树构建结果如图2所示。
NCBI数据库中BLASTp同源性分析表明,rAgaN3序列与已知琼胶酶序列agaraseMicrobulbiferthermotolerans(BAD29947.1)、agaraseMicrobulbiferthermotolerans(3WZ1A)以及agaraseMicrobulbifersp. AG1(ALN70307.2)都有88%的序列相似性。
系统进化树节点上的数字是自展值,用来分析进化树分支可信度。0.05代表每100个氨基酸中5个氨基酸替换。由经过给定的repetitions(500次)重排构树打分后,每个分支对应一个数值。数值越高,表示分支的可信度越高,由此来确定进化远近程度。rAgaN3与Microbulbiferthermotolerans(3WZ1A)、Microbulbiferthermotolerans(BAD29947.1)的自展值均为 100。结合BLASTp同源性分析,基本可以确定该蛋白来自Microbulbiferthermotolerans。
2.2 蛋白质一级结构预测
2.2.1 蛋白质理化性质分析
通过ProtParam预测蛋白rAgaN3的理化性质,结果见表1。

图 1 rAgaN3 基因序列和氨基酸序列Fig.1 rAgaN3 gene sequence and amino acid sequence
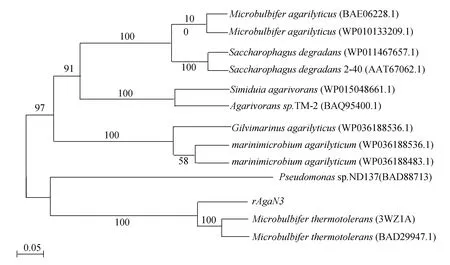
图 2 系统进化树Fig.2 Phylogenetic tree
2.2.2 亲疏水性分析
蛋白质内部亲疏水性氨基酸的组成影响蛋白质折叠的程度,蛋白质的折叠情况可利用亲水性分布图来反映。用ProtScale 计算出蛋白质亲疏水性分布图,用Hphob. / Kyte & Doolittle 标度,各个氨基酸打分分值见表2。
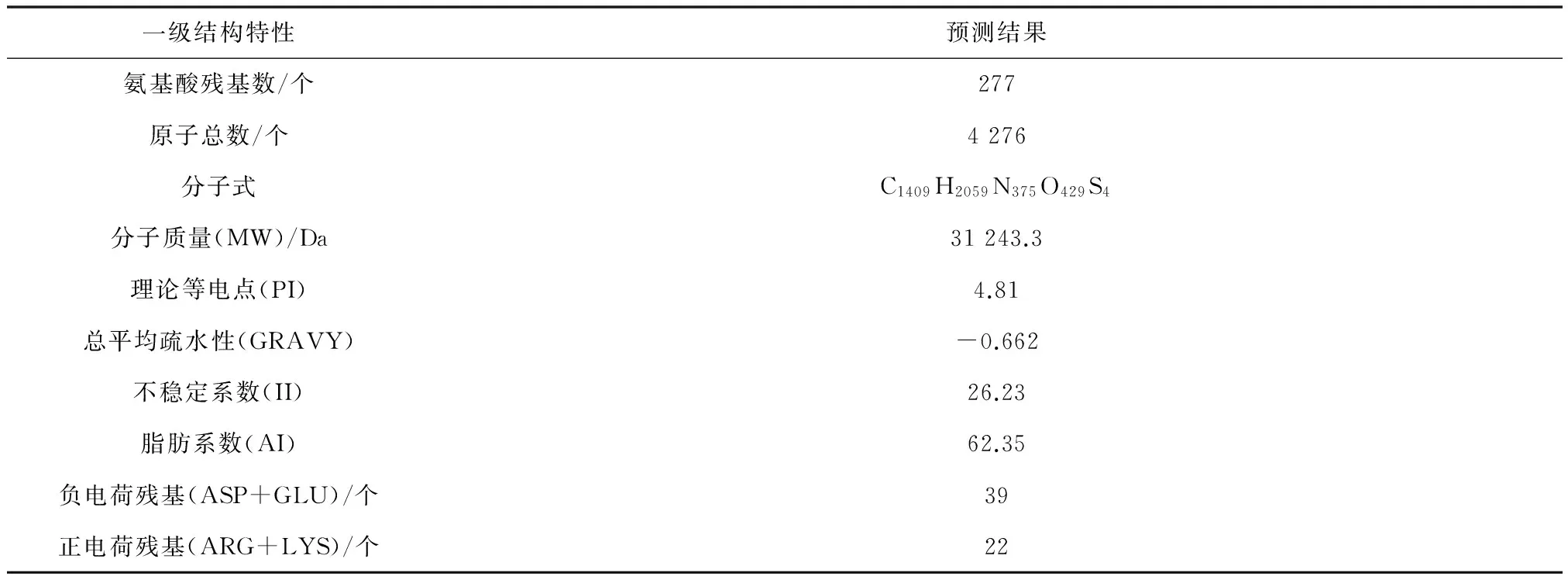
表 1 rAgaN3 理化性质分析表Table 1 Physical and chemical property analysis of rAgaN3
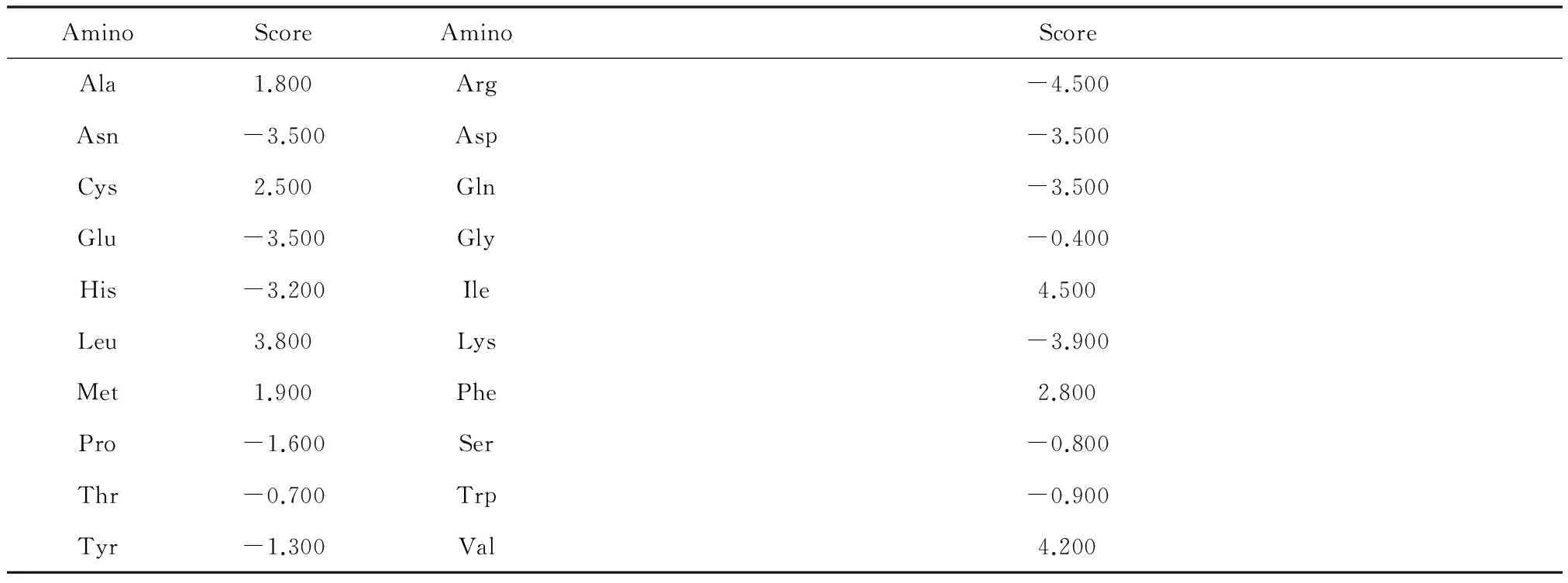
表 2 氨基酸分值表Table 2 Amino acid score
分析亲水区和疏水区,如图3所示。

图 3 琼胶酶氨基酸序列疏水性亲水性预测Fig.3 Predicted hydrophobicity/hydrophilicity of the amino acid sequence of agarase
横坐标表示氨基酸位置,纵坐标表示标度得分,Score小于0表示亲水,大于0表示疏水。图中显示亲水性值峰明显多于疏水性值峰,这些亲水性值峰区域常富集富集亲水性氨基酸,同时也是蛋白质进化中氨基酸插入的主要位点[20],推测此蛋白为亲水性蛋白,蛋白可溶于水。
2.2.3 跨膜区预结构预测与信号肽分析
蛋白质的跨膜区域是指蛋白在膜内与细胞膜膜脂相结合的部分,TMHMM是一种基于隐马尔可夫模型的跨膜螺旋预测算法,TMHMM的预测结果如图4所示。
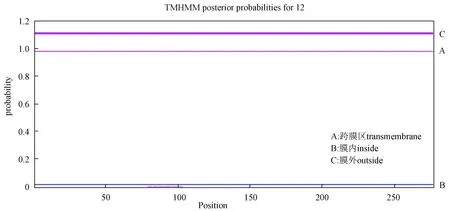
图 4 琼胶酶跨膜区预测Fig.4 Predicted transmembrane domain of agarase
TMHMM预测rAgaN3跨膜螺旋区域为零,说明该蛋白不是跨膜蛋白,推测该蛋白在细胞内合成之后不能立即分泌到胞外行使功能。
信号肽是将细胞内新合成的蛋白质引导分泌至胞外的短肽链,一般长度为5~30个氨基酸。SignalP是基于神经网络网络算法,预测给定氨基酸序列中潜在的信号肽剪切位点[14]。SignalP 4.1预测信号肽位置,结果如图5。

图 5 信号肽预测Fig.5 Signal P prediction of agarase
图中C-score是信号肽酶切位点值,一般剪切位点处的C-score最高;S-score是信号肽值,一般信号肽区域的S-score最高;Y-score是综合得出的剪切位点分值。预测结果显示mean S score为0.052,远小于分泌型蛋白标准0.5,结合图13,可以认为此蛋白没有信号肽。综合跨膜区域和信号肽预测,蛋白rAgN3为胞内蛋白,需要在菌体裂解之后才能在胞外行使其功能。
2.3 蛋白质二级结构预测
蛋白质二级结构包括螺旋(Helix)、折叠(Strand)、无规则卷曲(Coils)以及模体(motif)等组件。
2.3.1 PSIPRED预测蛋白二级结构
PSIPRED通过PSI-BLAST 搜索同源序列预测蛋白质二级结构,由于采用严格的交叉验证,使预测平均准确率高达80%。PSIPRED预测蛋白rAgaN3的二级结构结果如图6所示。

图 6 琼胶酶二级结构预测Fig.6 Predicted secondary structure of agarase
AA代表了目标蛋白质的氨基酸序列。Pred 代表了二级结构以及相应的图例(H:Helix;C:Coil;E:Strand)。Conf 数值在1~9 变化,数值越高,置信度越高。由PSIPRED预测结果可知 :rAgaN3二级结构主要由折叠和卷曲结构构成,其中含有15个折叠结构,其余为卷曲结构。
2.3.2 模体搜索
模体(motif)表示蛋白质中具有特定空间构象和特定功能的结构成分。用BLAST在默认的情况下进行了CD 搜索,向BLASTp提交序列后,获得的结果如图7所示。

图 7 模体结构Fig.7 Motif structure
该琼胶酶属于糖苷水解类16家族(GH16),属于LamG超家族;有11个活性位点分布于75~262氨基酸之间;有3个催化残基,3个Ca2+结合位点,但都在活性区域以外,推断Ca2+对琼胶酶活性可能无太大的促进作用。
2.4 结构域与功能分析
2.4.1 结构域分析
结构域是蛋白质序列功能、结构和进化的单元,通常由50~300 个氨基酸组成,有空间构象特异性。SMART预测结果如图8:该蛋白序列56~267 位氨基酸对应于Glyco_hydro_16 的结构域。

图 8 琼胶酶结构域Fig.8 Structural domain of agarase
Glyco_hydro_16在PFAM 数据库的编号为PF00722,进行查看和分析。糖苷水解酶16家族是糖苷水解酶的一个家族。糖苷水解酶(EC 3.2.1)是一类广泛存在的酶系,水解两个或以上的糖类。
查询CAZY数据库可知,糖苷水解酶16家族由许多具有已知活性的酶组成,包括: 葡聚糖酶、琼胶酶、透明质酸酶、半乳糖苷酶、卡拉胶酶、木聚糖酶等。
2.4.2 PredictProtein预测蛋白信息
欧洲分子生物学实验室提供的蛋白质序列和结构预测服务网站 PredictProtein,可以得到蛋白质多序列比对、低复杂区、溶剂可及性、跨膜螺旋、卷曲螺旋区、核定位信号及二级结构等信息。其中跨膜螺旋、二硫键、结合位点预测结果分别见图9、图10、图11。

图 9 跨膜螺旋区域预测Fig.9 Predicted transmembrane domain

图 10 二硫键预测Fig.10 Predicted disulfide bonds
PredictProtein预测得到:rAgaN3 无跨膜螺旋区域,与TMHMM预测结果一致。
PredictProtein预测得到:rAgaN3 无二硫键结构。

图11 结合位点预测Fig.11 Predicted binding sites
PredictProtein预测得到:rAgaN3 存在14 个蛋白质结合位点以及2 个多核苷酸结合位点。
PredictProtein预测分子功能本体论如表3所示。
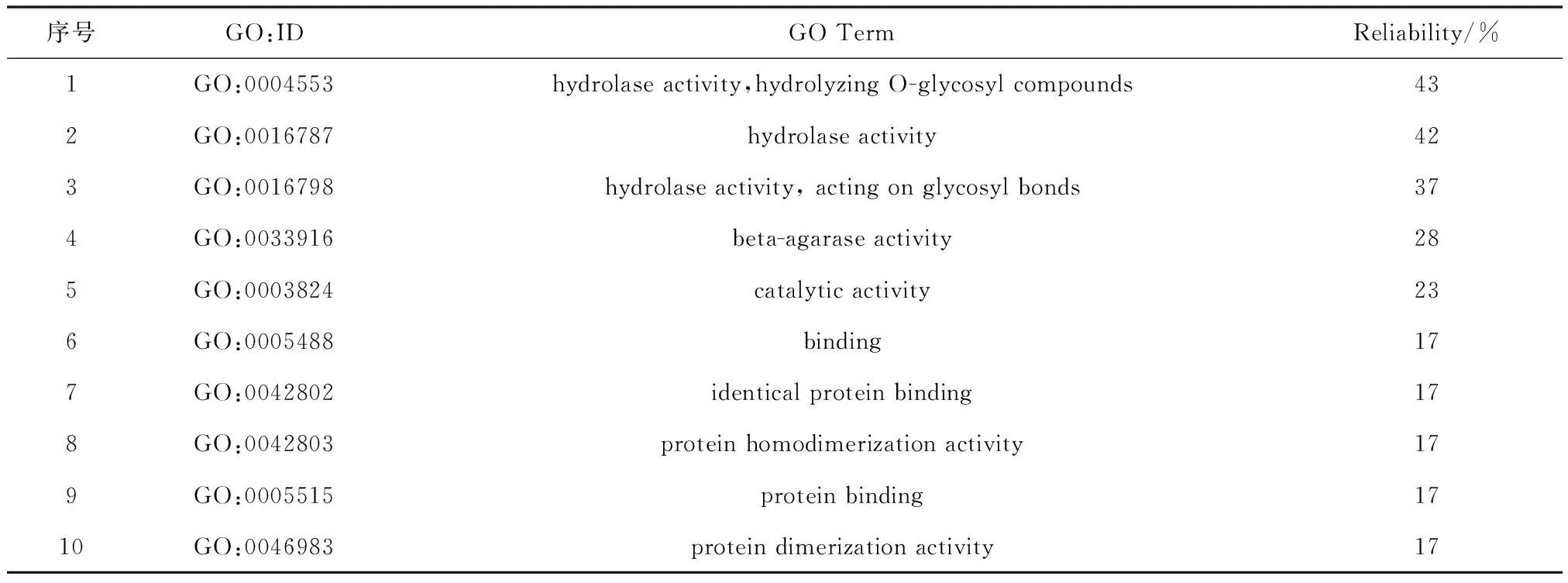
表 3 分子功能本体论Table 3 Molecular function ontology
分子功能本体论表明:该蛋白可能具有水解酶活性,水解O-糖苷化合物(GO:0004533);水解酶活性,作用于糖苷键(GO:0016798);β-琼胶酶活性(GO:0033916);催化活性(GO:0003824)。可靠性分别为43%、42%、37%,28%和23%。可以推测该蛋白是一种糖苷水解酶,与结构域分析该酶属于糖苷水解酶GH16结果一致。
PredictProtein生化过程本体论如表4所示。

表 4 生化过程本体论Table 4 Biological process ontology
生化过程本体论表明:该蛋白参与新陈代谢过程(GO:0008152),最初的新陈代谢过程(GO:0044238),糖类代谢过程(GO:0005975)和有机物质的代谢过程(GO:0071704)。可靠性分别为35%、27%、27%和27%,可以推测该蛋白参与体内新陈代谢。
2.4.3 亚细胞定位及导肽预测分析
用Target1.1进行亚细胞定位及导肽分析,结果如表5。
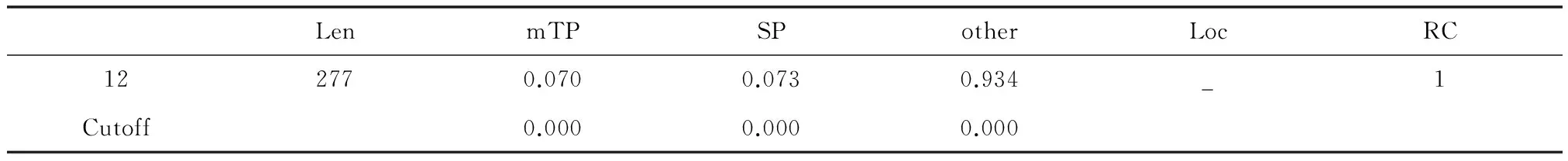
表 5 琼胶酶亚细胞定位Table 5 Subcellular location of agarase
Target1.1预测结果显示:该蛋白质氨基酸序列长277 个氨基酸,存在线粒体目标肽(mTP)的可能性为7.0%,存在信号肽(SP)的可能性为7.3%,其他导肽或无导肽的可能性为93.4%。目的蛋白rAgaN3 的分泌途径为—型,即定位在其他细胞器,没有剪切位点的序列,可靠性级别为1级。与跨膜螺旋区域、信号肽预测结果相匹配。
2.5 蛋白质三级结构预测
2.5.1 同源建模
SWISS-MODEL是目前应用最广泛的在线同建模网站,利用同源建模的方法实现对未知结构的序列的预测。在PDB数据库中找到与琼胶酶rAgaN3 的近同源蛋白3wz1A(MicrobulbiferthermotoleransJAMB-A94 GH16家族,β-琼胶酶)作为模板。rAgaN3与3wz1A的蛋白序列比对如图12所示。
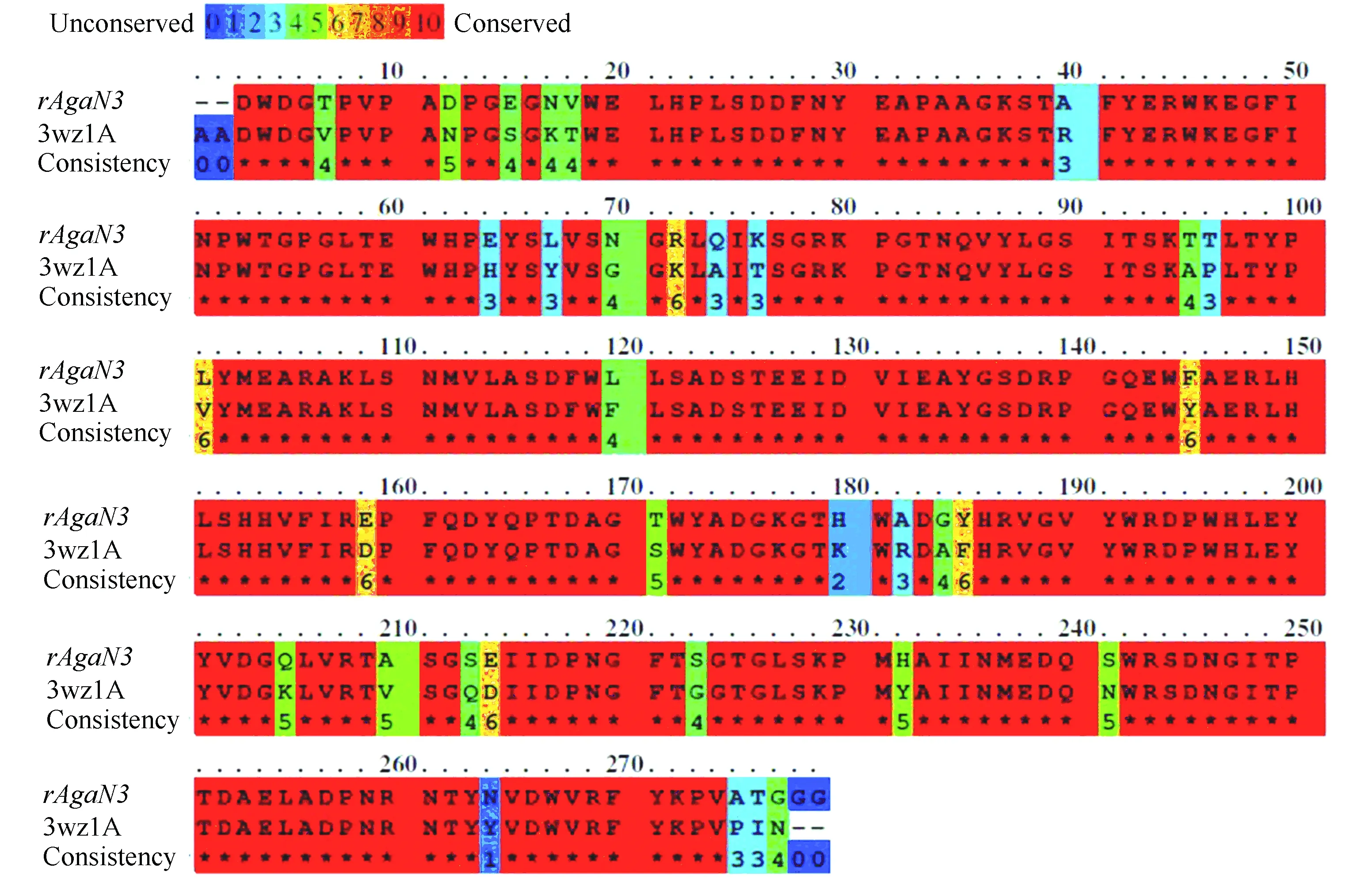
图 12 rAgaN3 与3wz1A 蛋白的序列比对Fig.12 Sequence alignment of rAgaN3 and 3wz1A protein
由图12可以看出,rAgaN3与模板蛋白序列大部分氨基酸是极保守的,相似程度达到88%,用3wz1A作为同源建模模板准确性较高。用SWISS-MODEL预测得到三维结构,序列从N 端到C 端分别用绿色到红色表示(见图13)。可以看出,琼胶酶rAgaN3的主要由β-折叠构成,整体结构类似“三明治”状,由15个折叠结构组成,与Allouch等[21]报道过的GH16家族的琼胶酶β-jelly-roll结构较为类似。
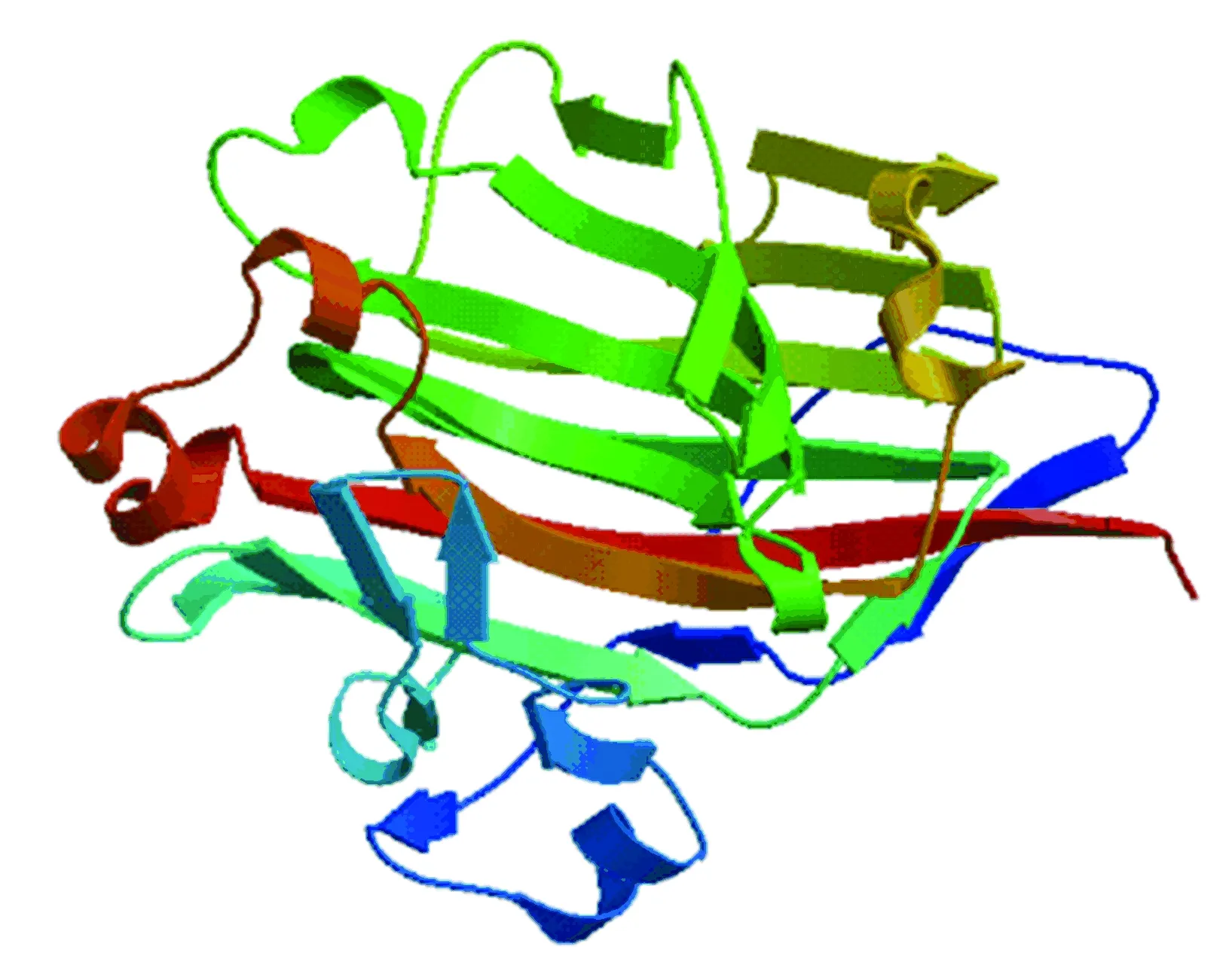
图 13 琼胶酶三级结构Fig.13 Tertiary structure of agarase
2.5.2 结构合理性评价
用SAVES服务器对结构进行PROCHECK评价,PROCHECK程序不考虑能量,只检测结构中的残基之间角度是否合理,生成Ramachandran plot。检测结果见图14。
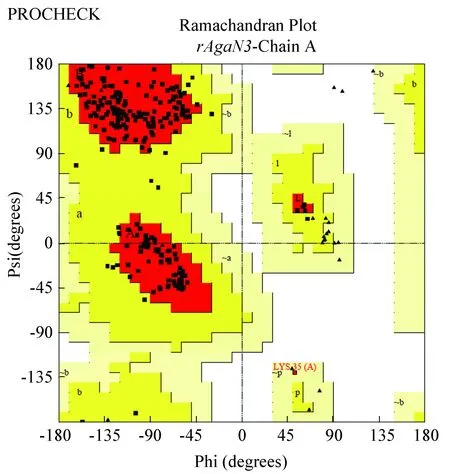
图14 拉式图Fig.14 Ramachandran plot
PROCHECK显示氨基酸残基核心区:91.3%,允许区:8.3%,大致允许区:0.4%,禁阻区:0%,位于可接受区的达到了100%,一般位于可接受区的氨基酸残基大于90%可以认为蛋白结构合理。rAgaN3建模得到的结构符合立体化学的原则,模型各残基之间的角度很合理。
PROSA程序是检测蛋白结构能量常用的工具,反映的是结构中残基之间的能量是否合理。得到的结果一般为负值。程序评价结果见图15。

图15 Z-score图Fig.15 Z-score of rAgaN3
本实验的Z-score为-7.04(图中黑点所示),蓝色部分为所有已解析的晶体结构的Z-score分布情况。由图可知,rAgaN3的Z-score位于中心的位置,说明该结构在能量上是十分合理的。综合Ramachandran plot和Z-score的结果分析,SWISS-MODEL构建出的琼胶酶结构是相当可靠的。
3 结论
重组琼胶酶rAgaN3理论分子量为31.243 kDa,理论等电点为4.81,脂肪系数为62.35,不稳定系数为26.23,小于40,说明蛋白较稳定,总平均疏水性为-0.662,小于0,预测为亲水性蛋白;二级结构预测得到:该蛋白无螺旋结构,有15 个折叠结构,其余均为卷曲结构。无信号肽,无跨膜区域也没有二硫桥。根据序列分析表明,rAgaN3属于糖苷水解酶GH16 家族,为β-琼胶酶。采用序列比对算法搜索蛋白质结构PDB 数据库,找到与琼胶酶rAgaN3的近同源蛋白3wz1A(MicrobulbiferthermotoleransJAMB-A94 GH16 家族,β-琼胶酶),序列相似性达到88%。运用同源建模法,使用SWISS-MODEL 构建重组琼胶酶rAgaN3的三维结构模型,可以认为此琼胶酶是典型的GH16家族琼胶酶结构,并用PROCHECK和PROSA进行了结构检验。
本研究从琼胶酶的序列出发,对序列中包含的生物信息进行了系统的研究,预测了琼胶酶rAgaN3的基本理化性质,为理解琼胶酶特性提供了参考。后续工作可以本文预测信息为基础,将基因在大肠杆菌表达系统和毕赤酵母表达系统中进行异源表达,并对重组琼胶酶的酶学性质进行测定,以验证生物信息学预测结果。用高同源性的模板同源建模得到的较可靠的蛋白质三维结构,为理解琼胶酶的结构特性以及催化机制打下了良好的基础。并且可以从蛋白质的三维结构出发,通过分子对接、分子动力学模拟等方法,以提高比活力以及改善酶学性质为目标,预测潜在的突变位点并进行定点突变实验,深入研究琼胶酶结构与功能的关系。
References)
[1]梅建凤, 李莎, 茅鹤婷, 等. 一株产琼胶酶海洋细菌的分离与鉴定[J]. 海洋科学, 2014,38(2): 71-75.DOI: 10.11759/hykx20130502003.
MEI Jianfeng,LI Sha,MAO Heting,et al. Isolation and identification of a marine bacteria producing agarase[J]. Marine Sciences, 2014, 38(2): 71-75. DOI: 10.11759/hykx20130502003.
[2]POTIN P, RICHARD C, ROCHAS C, et al. Purification and characterization of the α-agarase fromAlteromonasagarlyticus(Cataldi) comb. nov., strain GJ1B[J]. European Journal of Biochemistry,1993,214(2): 599-607. DOI: 10.1111/j.1432-1033.1993.tb17959.x.
[3]KIRIMURA K, MASUDA N, IWASAKI Y, et al. Purification and characterization of a novel beta-agarase from an alkalophilic bacterium,Alteromonassp. E-1.J[J].Bioscience and Bioengineering,1999, 87(4): 436-441.DOI: 10.1016/S1389-1723(99)80091-7.
[4]USOV A I, MIROSHNIKOVA L I. Isolation of agarase fromLittorinamandshuricaby affinity chromatography on Biogel A[J]. Carbohydrate Research, 1975, 43(1): 204-207. DOI:10.1016/S0008-6215(00)83989-0.
[5]USOV A I, MARTYNOVA M D.Detection of agarase in molluscs of the genusLittorina[J]. Doklady Akademii Nauk SSSR, 1970, 194(2): 455-457.
[6]胡晓珂,江晓路,管华诗.海藻多糖降解酶的性质和作用机理[J].微生物学报, 2001,41(6):762-766.
HU Xiaoke, JIANG Xiaolu, GUAN Huashi. Properties and mechanisms of marine polysaccharisases[J].Acta Microbiologica Sinica, 2001(6): 762-766.
[7]MORTICE L M, MCLEAN M W, LONG W F, et al.Porphyran primary structure. An investigation using beta-agarase I from Pseudomonas atlantica and 13C-NMR spectroscopy[J]. European Journal of Biochemistry, 1983, 133(3): 673-684. DOI: 10.1111/j.1432-1033.1983.tb07516.x.
[8]SUGANO Y, TERADA I, ARITA M, et al.Purification and characterization of a new agarase from a marine bacterium,Vibriosp. strain JT0107[J]. Applied and Environmental Microbiology,1993,59(5): 1549-1554.
[9]BUTTNER M J, FEARNLEY I M, BIBB M J.The agarase gene (dagA) of Streptomyces coelicolor A3:nucleotide sequence and transcriptional analysis[J]. Molecular and General Genetics MGG, 1987,209(1): 101-109. DOI: 10.1007/BF00329843.
[10]TAKAGI E, HATADA Y, AKITA M, et al. Crystal structure of the catalytic domain of a GH16 β-agarase from a deep-sea bacterium, microbulbifer thermotolerans JAMB-A94[J]. Bioscience, Biotechnology,s&sbiochemistry, 2014, 79(4): 1-8.DOI:10.1080/09168451.2014.988680.
[11]GASTEIGER E, HOOGLAND C, GATTIKER A, et al. Protein identification and analysis tools on the ExPASy Server[J]. Proteomics Protocols Handbook, 2005, 112(112): 571-607.DOI:10.1385/1-59259-890-0:571.
[12]熊伟, 杨勇琴, 张海洋,等.人线粒体转录终止因子1(hMTERF1)蛋白的生物信息学分析[J]. 生物信息学, 2015, 13(1): 23-30.DOI:10.3969/j.issn.1672-5565.2015.01.01.
XIONG Wei,YANG Yongqin,ZHANG Haiyang,et al. Bioinformatic analysis of human mitochondrial transcription termination factor 1(hMTERF1)[J]. Chinese Journal of Bioinformatics,2015, 13(1): 23-30. DOI:10.3969/j.issn.1672-5565.2015.01.01.
[13]周雅, 高贝, 张道远. 齿肋赤藓早期光诱导蛋白ELIPs的生物信息学分析[J]. 生物信息学, 2014, 12(4): 233-241.DOI:10.3969/j.issn.1672-5565.2017.04.01.
ZHOU Ya,GAO Bei,ZHANG Daoyuan. Bioinformatic analysis of early light-induced protein (ELIPs)inSyntrichiacaninervis[J]. Chinese Journal of Bioinformatics,2014, 12(4): 233-241. DOI:10.3969/j.issn.1672-5565.2017.04.01.
[14]PETERSEN T N, BRUNAK S, VON H G, et al. SIGNALP 4.0: discriminating signal peptides from transmembrane regions[J]. Nature Methods, 2010, 8(10): 785-786. DOI: 10.1038/nmeth.1701.
[15]SCHULTZ J, MILPETZ F, BORK P, et al. SMART, a simple modular architecture research tool:Identification of signaling domains[J]. Proceedings of the National Academy of Sciences, 1998,95(11): 5857-5864. DOI: 10.1073/pnas.95.11.5857.
[16]LOMBARD V, GOLACONDA R H, DRULA E, et al. The carbohydrate-active enzymes database (CAZy) in 2013.[J]. Nucleic Acids Research, 2014, 42(Database issue): 490-495. DOI: 10.1093/nar/gkt1178.
[17]ARNOLD K, BORDOLI L, KOPP J, et al.The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling[J]. Bioinformatics, 2006, 22(2): 195-201. DOI: 10.1093/bioinformatics/bti770.
[18]LASKOWSKI R A, RULLMANNN J A, MACARTHUR M W, et al. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR[J]. Journal of Biomolecular Nmr, 1996,8(4): 477-486.
[19]WIEDERSTEIN M, SIPPL M J.ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins[J]. Nucleic Acids Research, 2007, 35(Web Server issue): 407-410. DOI: 10.1093/nar/gkm290.
[20]王明强, 高贝, 张道远. 2个银叶真藓HSP70序列的生物信息学相关分析[J]. 生物信息学, 2015(1): 1-8.DOI:10.3969/j.issn.1672-5565.2015.01.01.
WANG Mingqiang,GAO Bei,ZHANG Daoyuan.Bioinformatics relevant analysis of two heat shock protein 70 sequence fromBryumargenteum[J]. Chinese Journal of Bioinformatics,2015, 13(1): 1-8. DOI:10.3969/j.issn.1672-5565.2015.01.01.
[21]ALLOUCH J, JAM M, HELBERT W, et al. The three-dimensional structures of two β-agarases[J]. Journal of Biological Chemistry, 2003, 278(47): 47171-47180. DOI: 10.1074/jbc.M308313200.
Bioinformatics analysis of the recombinantrAgaN3 gene of agarase
XIE Yong,HONG Xiaokun,YAN Renxiang,LIN Juan*
(FujianProvincialKeyLaboratoryofMarineEnzymeEngineering(FuzhouUniversity),Fuzhou350116,China)
In this paper, the agarase enzyme gene namedrAgaN3 is analyzed which is cloned fromMicrobulbifersp. BN via various bioinformatics methods. The results show that the calculated molecular mass of reorganization of agaroserAgaN3 is 31.243 kDa, the theoretical isoelectric point is 4.81, 26.23 of the nstability coefficient, 62.35 of the fat index, and minus 0.662 of the average hydrophobic coefficient, and there is no transmembrane domain and no signal peptide. By analyzing the secondary structure of protein, we can find there are 15 β-sheet structures, other coiled structures, and no helical structure. According to the analysis of sequence similarities, we can know rAgaN3 is a β-agarase, belongs to glycoside hydrolase GH16 family. It is templated by a homologous protein 3wz1A (88 homology), sets up tertiary structure of protein by creating homology modeling and tests its structure by Ramachandran Plot and PROSA. The bioinformatics analysis ofrAgaN3 gene of Agarase can provide a guidance to heterologous expression of agarase and lay good foundations for the site-directed mutagenesis of agarase and comprehensive study on the relationship between structure and function.
Agarase; Gene analysis; Bioinformatics; Protein structure
2016-08-20;
2016-10-18.
福建省企业技术创新项目(闽经信投资[2015]205号);福州市科技计划项目(No.2016-G-42)。
谢勇,男,硕士研究生,研究方向:微生物学;E-mail:1148494580@qq.com.
*通信作者:林娟,女,教授,博士生导师,研究方向:微生物学及生物化学与分子生物学;E-mail: ljuan@fzu.edu.cn.
10.3969/j.issn.1672-5565.2017.01.201608003
Q55
A
1672-5565(2017)01-016-11
猜你喜欢
河北科技大学学报(2023年5期)2023-11-09 01:44:44
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
现代畜牧科技(2021年4期)2021-07-21 06:12:50
生物学通报(2019年3期)2019-02-17 18:03:58
中国博物馆(2018年2期)2018-12-05 05:28:50
浙江农业学报(2017年3期)2017-04-08 02:39:02
华东理工大学学报(自然科学版)(2015年4期)2015-12-01 04:00:23
华东理工大学学报(自然科学版)(2015年4期)2015-12-01 04:00:23
现代检验医学杂志(2015年4期)2015-02-06 02:01:55
