
基于微量DNA样本的简化甲基化文库构建方法研究
2017-04-10 06:10:57魏冬凯徐康萍
生物信息学 2017年1期
魏冬凯,南 蓬,徐康萍,裘 锋,3*
(1. 苏州贝斯派生物科技有限公司 江苏 苏州 215123;2. 复旦大学生命科学学院 上海 200438;3.上海生物信息技术研究中心 上海 201203)
基于微量DNA样本的简化甲基化文库构建方法研究
魏冬凯1,2,南 蓬2,徐康萍1,裘 锋1,3*
(1. 苏州贝斯派生物科技有限公司 江苏 苏州 215123;2. 复旦大学生命科学学院 上海 200438;3.上海生物信息技术研究中心 上海 201203)
RRBS(简化甲基化测序)是一种有效研究DNA甲基化状态的测序方案。文库构建是该方案中最关键的实验步骤之一,RRBS文库构建往往因为酶切产物少,文库构建步骤繁多等因素,导致DNA起始量需要1 ug以上。然而很多来源于人的样本,如冰冻组织穿刺、石蜡切片、显微微切割等,都往往只能获得100 ng以下的DNA,无法满足常规RRBS文库构建的起始DNA总量要求,从而大大限制RRBS技术的应用范围。本研究主要探讨并设计了基于微量DNA样本(<100 ng)的RRBS文库构建策略,主要通过优化MspI酶切条件、DNA片段大小筛选、缩减反应步骤及减少DNA转移次数等技术手段,大大提高了DNA回收率,进而建立了2种有效使用微量DNA样品的RRBS文库构建方案(即EA-Method和WB-Method方案)。EA-Method方案在处理100 ng左右的DNA时,有经济、快速、高效的特点,但文库质量略差于WB-Method方案;WB-Method方案可将DNA起始量降低至10 ng,并且文库质量高。为验证WB-Method方法的可靠性,研究人员选取了3种志愿者样本(全血和口拭子),采用WB-Method同时进行常量和微量RRBS文库构建,并进行Illumina Hiseq平台测序,数据通过生物信息分析得到微量RRBS检测的CpG,CHG,CHH中C碱基的甲基化比例与常量RRBS结果一致。同时,该方法可以大大降低DNA损耗及损伤,因而该方法可将RRBS技术应用到更广泛的样本类型中去,有效拓展RRBS的研究领域。
二代测序;RRBS;简化甲基化;EA-M;WB-M
表观遗传学是指在不改变DNA 序列的条件下所发生的可遗传基因表达的变化[1].表观遗传学主要包括DNA 甲基化、组蛋白修饰和染色质结构.脊椎动物中,DNA 甲基化表现为在DNA 甲基化转移酶(DNA methyltransferase,DNMT)作用下,甲基基团合成到5′-CpG-3′中胞嘧啶的第五位碳原子上[2].正常情况下,人类基因组“垃圾”序列的CpG二核苷酸相对稀少,并且总是处于甲基化状态。非甲基化的CpG不是均匀分布,而是呈现局部聚集倾向,形成一些GC含量较高、CpG双核苷酸相对聚集的区域,即CpG岛[3].与之相反,人类基因组中大小为100~1000 bp 左右且富含CpG二核苷酸的CpG岛则总是处于未甲基化状态,并且与56%的人类基因组编码基因相关[4]。在正常人的基因组DNA中,约有3%~6%的胞嘧啶是甲基化的[5]. 根据Gardiner-Garden 等[6]的定义,CpG 岛是一段长度不小于200 bp、GC 含量不小于50%、CpG 含量与期望含量之比不小于0.6 的区域.由于该定义将一些重复片段也包含其中,Takai 和Jones[7]将CpG 岛重新定义为长度不小于500 bp、GC 含量不小于55%、CpG 含量与期望含量之比不小于0.65的区域.据统计,多于50%的基因的启动子区含有CpG 岛[8-9].在早期的认识中,CpG 岛都是非甲基化的,但是随着研究的不断深入,人们发现在印迹基因、失活的X 染色体[10]甚至是正常的体细胞中都存在甲基化的CpG 岛[11].部分CpG 岛的异常甲基化常常伴随着癌症等疾病的发生[12]。高通量测序技术堪称测序技术发展历程的一个里程碑,该技术可以对数百万个DNA 分子同时进行测序。这使得对一个物种的转录组和基因组进行全景式分析成为可能,因此也称其为深度测序(Deep sequencing)[13]或下一代测序技术(Next generation sequencing,NGS)[14]。新一代高通量测序技术使得基因组整体水平高精度的甲基化检测成为现实。目前,高通量测序技术已经广泛应用于拟南芥、人、水稻和家蚕等生物DNA甲基化的研究,并取得了广泛的研究成果[15-18]。
研究DNA 甲基化的高通量测序技术方法主要包括:全基因组重亚硫酸盐测序(Bisulfite sequencing,Bi-seq) 和简化表达重亚硫酸盐测序 (Reduced Representation Bisulfite Sequencing, RRBS)、甲基化DNA 免疫共沉淀测序(Methylated DNA immuno precipitation sequencing, MeDIP-seq)、甲基化DNA 富集结合高通量测序(Methylated DNA binding domain sequencing, MBD-Seq)等。目前,重亚硫酸盐修饰被公认为DNA 甲基化检测的“金标准”。Bi-seq和RRBS技术均是基于上述修饰。全基因组Bisulfite 测序技术可以得到单碱基分辨率的全基因组甲基化图谱。这一技术最初被Cokus等[15]应用在植物拟南芥的全基因甲基化图谱分析上(借助了Solexa 测序技术)。2009年Lister 等[19]利用全基因组Bisulfite 测序发表了第一篇人类的全基因组甲基化图谱( 利用了Solexa 测序),随后该技术开始在人类疾病,如肥胖症的研究中得以应用[20]。基于对黄种人的外周血单核细胞DNA 样品进行Bisulfite 处理后,结合Solexa 测序技术进行深度测序,2010年Li等[21]成功绘制出了“炎黄一号”高精确度的全基因组甲基化图谱。然而,重亚硫酸盐转化全基因组法高昂的测序成本,在一定程度上限制了该技术的广泛应用。
简化表达亚硫酸盐测序(RRBS),又称基于酶切消化的重亚硫酸盐测序, 是一种简便、高效、经济的DNA甲基化研究方法,通过酶切富集启动子及CpG岛区域,并进行Bisulfite测序,同时实现DNA甲基化状态检测的高分辨率和测序数据的高利用率。2005年Meissner 等[22]最初结合Sanger 测序发明了该项技术,并将其成功应用于鼠胚胎干细胞去甲基转移酶前后的甲基化谱的检测。接着Meissner领导的研究小组通过改用MspI 酶切来富集小鼠全基因组近90%的CpG岛片段,结合Illumina 公司的高通量测序技术,建立并完善了适用于哺乳动物全基因组甲基化测序分析的方法[23]。2010年该课题组又发表了一篇关于RRBS的文章,在其先前的工作基础上进一步优化RRBS 技术并挖掘其在临床应用上的潜力[24]。2012年华大基因研究院与深圳大学、中山大学及华南科技大学合作发表了RRBS与Bisulfite 测序的比较方法学文章[25]。Wang等[26]发现双酶切(MspI,ApeKI)可以提高RRBS建库成功率,并且提高覆盖率。
目前RRBS构建测序文库时所需DNA出发量一般大于100 ng[27],某些样本甚至需要到达1 μg以上[28-29]。然而很多情况下实验样本很珍贵,如FFPE样本或者单细胞样本,这样对RRBS建库的要求和难度都大幅度提高,目前对于少于100 ng的样本进行RRBS建库缺乏有效的技术方法,本实验对从微量DNA样品出发构建RRBS文库进行了较为细致的研究,并试图找出并建立一种更简便高效的方法。这对于拓宽简化甲基化测序的应用领域有着重大意义。
1 材料与方法
1.1 材料
本实验所使用的gDNA来源为实验室自传代人类红白血病细胞系K562细胞,细胞采用悬浮培养至密度80%(K562细胞作为一种常见的研究用细胞系,具有一定代表性,并为保证细胞状态最佳,所以将细胞培养至密度80%左右。),培养皿大小100 mm,培养基选用DMEM+10%FBS(胎牛血清),为保证gDNA来源保持一致,本实验所用DNA采用一次性提取3个培养皿的混合K562细胞所得。
本实验中,用于验证WB-Method可靠性的人源样本来源于3位志愿者,常量样本采用静脉血200 μL,微量样本采用少量口腔上皮细胞(为验证WB-M方案应用至真实人源样本的可靠性,所以验证实验采用志愿者自采集样本。)。
1.2 gDNA提取
gDNA提取方法采用蛋白酶K消化法,将细胞收集至15 mL离心管中,加入3 mL PBS缓冲液重悬细胞后,依次加入500 μL蛋白酶K(20 mg/mL)、7 mL 裂解液(SDS、EDTA、Tris-HCl),放置56 ℃孵育4 h后,1∶1(V/V)加入酚氯仿异戊醇(25∶24∶1)溶液,充分混匀后离心取上清,再按1∶1加入氯仿异戊醇(24∶1)溶液,充分混匀后离心取上清,加入0.8倍体积异丙醇(-20 ℃预冷)充分混匀后,可见白色团状沉淀,使用枪头将沉淀捞出,并使用70%乙醇漂洗沉淀2次,干燥挥发乙醇3 min后,使用Qiagen EB Buffer溶解沉淀。提取的DNA采用Qubit,NanoDrop和电泳进行质检。
静脉血gDNA提取方法使用QiaAmp Blood Mini Kit,按照操作手册进行DNA提取,大致方法如下:将200 μL静脉血置于冰水混合物中融化后加入20 μL蛋白酶K和600 μL AL缓冲液,混匀后置于56 ℃水浴15~30 min,裂解产物加入1倍体积乙醇充分混匀后过离心柱,所有产物过离心柱后依次加入AW1、AW2缓冲液洗涤离心柱2次,空气干燥2~3 min,加入AE缓冲液洗脱DNA,提取的DNA采用Qubit,NanoDrop和电泳进行质检。
1.3 MspI酶切条件优化
本实验使用MspI限制性内切酶(NEB,20 U/μL)对gDNA进行酶切处理(因MspI酶切位点CCGG与人类CpG Island序列相似,并且该酶对甲基化区域不敏感,可以最大限度的富集CpG Island区域。),得到有效的简化末端。因MspI酶对温度不敏感,所以工作中只对MspI酶的用量和酶切时间进行条件优化实验。优化实验材料使用8 μg gDNA均分为7份,保证每份gDNA>1 μg,分别对每份gDNA,在37 ℃下采用不同时间和不同酶用量进行酶切,酶切体积50 μL,酶切完成后,65 ℃变性20 min,取25 μL酶切产物加入6X Loading Dye 5 μL,点2% 琼脂糖凝胶 100 V 电泳2 h检测酶切效率并回收相应片段。
1.4 片段大小筛选方法优化
片段大小筛选采用2种方法进行,分别是琼脂糖凝胶电泳(加入Carrier DNA)(本实验中所用的Carrier DNA为完全未甲基化的大肠杆菌DNA,unmethylation DNA,Promega)和磁珠两步法筛选。
琼脂糖凝胶电泳(加入Carrier DNA)方案,采用1 μg gDNA MspI 酶切后并进行补平和连接测序接头所得的连接产物为材料,Carrier DNA采用unmethylation DNA (Promega)covaris打断至150~300 bp,在连接产物中加入200 ng,不加入Carrier DNA的做为对照,进行2%琼脂糖凝胶电泳,100 V 2h检测加入CarrierDNA的效果。
磁珠两步法筛选大小方案,方案原理基于AMpureXP Beads对PEG(MW 6 000)的终浓度敏感,从而影响结合DNA片段大小。本实验采用1 μg gDNA covaris打断至100~700 bp作为材料,不同PEG浓度进行片段大小筛选,并采用2%琼脂糖凝胶电泳和BioAnalyzer 2100(毛细管电泳)进行片段大小检测。
1.5 常规RRBS建库方法
常规RRBS建库方法 (Conventional Method, CO-M)(见图1(a)),采用1 μg,100 ng,50 ng,10 ng gDNA,加1 μL (20 U/μL)MspI(以下所有使用酶试剂及酶反应buffer,均使用NEB来源,除特殊标注外)酶切 37 ℃ 0.5 h,加入末端修复反应试剂(10× Ligase Buffer 10 μL, 10 nM dNTPs 4 μL, 10 mM ATP 2.5 μL, T4 DNA Polymerase(3 U/μL) 5 μL,T4 PNK(10 U/μL) 5 μL,Klenow Large Fragment(5 U/μL) 1 μL,并补水至100 μL,20 ℃ 孵育30 min,产物用QiaAmp MinElute PCR Kit(Qiagen)进行纯化并洗脱至30 μL,加入末端加A反应试剂(10mmol/L dATP 5 μL,Klenow Buffer 5 μL,5 U/μL Klenow exo-3 μL),补水至50 μL,37 ℃孵育30 min,产物用QiaAmp MinElute PCR Kit(Qiagen)进行纯化并洗脱至30 μL,加入测序接头连接反应试剂(甲基化处理的测序接头(Illumina)1 μL,T4 ligase (10 U/μL) 3 μL,10×T4 ligase buffer 5 μL),加水补充至50 μL,22 ℃孵育15 min,连接产物使用QiaAmp MinElute PCR Kit(Qiagen)进行纯化并洗脱至75 μL,重亚硫酸盐CT转化采用Zymo Gold Methylation Kit (Zymo Research),操作根据试剂盒说明书并将产物纯化至20 μL,加入6× loading dye (Life Technologies)4 μL,2%琼脂糖凝胶电泳回收150~350 bp片段,胶回收采用QiaAmp MinElute Gel Extraction Kit(Qiagen),回收至20 μL。加入文库扩增试剂(Illumina Primer Mix 5 μL,2× KAPA uracil+ PCR Ready Mix (KAPA Biosystem) 20 μL,反应程序95 ℃ 10 min,扩增循环(95 ℃ 30 s,62 ℃ 30 s,72 ℃ 30 s)(1 μg DNA起始采用 10个循环,100 ng DNA及以下采用15个循环),72 ℃ 4 min),得到的PCR产物采用QiaAmp MinElute PCR Kit(Qiagen)进行纯化至20 μL,Qubit测定文库浓度,BioAnalyzer 2100(Agilent)检测文库片段分布。
1.6 EA一步RRBS建库方法
EA一步建库方法(EA Method, EA-M)(见图1(b)),采用100 ng,50 ng,10 ng gDNA,加MspI(以下所有使用酶试剂及酶反应buffer,均使用NEB来源,除特殊标注外)1 μL酶切 37 ℃ 0.5 h,加入末端修复反应试剂(10× Ligase Buffer 2.5 μL, 10 nmol/L dNTPs 1 μL, 10 mmol/L ATP 2.5 μL, T4 DNA Polymerase (3 U/μL)2.5 μL,T4 PNK(10 U/μL) 2.5 μL, Taq Polymerase(10 U/μL) 1 μL),补水至25 μL,12 ℃15 min,37 ℃ 15 min,72 ℃ 20 min,产物不纯化,直接加入测序接头连接反应试剂(甲基化处理后的测序接头(Illumina)1 μL,T4 ligase 3 μL,T4 ligase buffer 1 μL,加水补充至35 μL),22 ℃孵育15 min,连接产物使用QiaAmp MinElute PCR Kit(Qiagen)进行纯化并洗脱至75 μL,重亚硫酸盐CT转化采用Zymo Gold Methylation Kit(Zymo Research) ,操作根据试剂盒说明书并将产物纯化至20 μL,加入6× loading dye(Life Technologies) 4 μL,2%琼脂糖凝胶电泳回收150~350 bp片段,胶回收采用QiaAmp MinElute Gel Extraction Kit(Qiagen),回收至20 μL。加入文库扩增试剂(Illumina Primer Mix 5 μL,2× KAPA uracil+ PCR Ready Mix (KAPA Biosystem) 20 μL,反应程序95 ℃ 10 min,扩增循环(95 ℃ 30 s,62 ℃ 30 s,72 ℃ 30 s)(1 μg DNA起始采用 10个循环,100 ng DNA及以下采用15个循环),72 ℃ 4 min),得到的PCR产物采用QiaAmp MinElute PCR Kit(Qiagen)进行纯化至20 μL,Qubit测定文库浓度,BioAnalyzer 2100(Agilent)检测文库片段分布。
1.7 磁珠包被RRBS建库方法
磁珠包被RRBS建库方法 (WB Method, WB-M)(见图 1(c)),采用100 ng,50 ng,10 ng gDNA,加1 μL MspI酶(以下所有使用酶试剂及酶反应buffer,均使用NEB来源,除特殊标注外) 37 ℃ 酶切过夜,加入末端修复试剂(10× Ligase Buffer 10 μL, 10 nmol/L dNTPs 4 μL, 10 mmol/L ATP 2.5 μL, T4 DNA Polymerase(3 U/μL) 5 μL,T4 PNK(10 U/μL) 5 μL, Klenow Large Fragment(5 U/μL) 1 μL),补水至100 μL,20 ℃ 孵育30 min,产物用150 μL Ampure XP Beads(Beckman)纯化,纯化步骤按照AmpureXP Beads说明书进行至挥发晾干残留乙醇,加EB Buffer(Qiagen)37 μL,室温放置1 min,无需去除磁珠,直接加入末端加A反应试剂(10 mmol/L dATP 5 μL,Klenow Buffer 5 μL,5 U/μL Klenow exo-3 μL),37 ℃孵育30 min,产物加入PEG/NaCl(20% PEG8000(Sigma), 9% NaCl(Sigma))溶液 50 μL,重悬磁珠,并按照AmPureXP Beads说明书进行至挥发晾干残留乙醇,加EB Buffer(Qiagen)41 μL,室温放置1 min,无需去除磁珠,加入测序接头连接反应试剂(甲基化处理后的测序接头(Illumina)1 μL,T4 ligase(10 U/μL) 3 μL,T4 ligase buffer 5 μL),加水补充至50 μL,22 ℃孵育15 min,产物加入PEG/NaCl(20% PEG8000, 9% NaCl)溶液 50 μL,重悬磁珠,并按照AmPureXP Beads说明书进行至挥发晾干残留乙醇,加EB Buffer(Qiagen)75 μL,室温放置1 min,无需去除磁珠,重亚硫酸盐CT转化采用Zymo Gold Methylation Kit 并纯化至20 μL(本步骤使用Zymo试剂盒自带纯化柱,将磁珠去除),产物采用AmpureXP Beads 二步筛选法筛选片段大小至150~350 bp之间,采用EB Buffer洗脱至20 μL,加入文库扩增反应试剂(Illumina Primer Mix 5μL,2× KAPA uracil+ PCR Ready Mix (KAPA Biosystem) 20 μL,反应程序95 ℃ 10 min,扩增循环(95 ℃ 30 s,62 ℃ 30 s, 72 ℃ 30 s)(1μg DNA起始采用 10个循环,100 ng DNA及以下采用15个循环),72 ℃ 4 min),得到的PCR产物采用AmpureXP Beads进行纯化至20 μL,Qubit测定文库浓度,BioAnalyzer 2100检测文库片段分布。

图1 三种RRBS文库构建方法流程图Fig.1 Three methods for RRBS library construction
1.8 WB-Method验证实验
本实验使用人来源样本3例,采用WB-Method分别进行微量(10 ng)RRBS和常量(1 μg)RRBS文库构建,并使用Illumina Hiseq 3000平台进行高通量测序,每个样本测序20 mol/L reads以上。生物信息分析使用Trim_galore进行低质量(Q<20)数据过滤和去重,bismark进行序列比对,参考基因组选用人类基因组hg19,并对甲基化频率分布、甲基化覆盖度、3种context(CpG、CHG、CHH)下C碱基甲基化比例进行统计。
2 结果
2.1 gDNA提取及质量测定
gDNA提取方法采用经典PK法(细胞和组织样本),全血采用QiaAmp使用Blood Mini Kit,Qubit测定浓度并计算K562 gDNA总量为386 μg(Qubit 2.0 HSDNA Assay),Nanodrop 2000检测gDNA纯度(OD260/OD280=1.91,OD260/OD230=2.12 ),0.6% 琼脂糖凝胶检测gDNA纯度,电泳条带清晰单一,无降解拖尾条带,片段均在40 kb以上(见图2)。人来源的每个常量样本均获得2 μg以上DNA(OD260/OD280=1.94,OD260/OD230=2.08),微量样本获得100 ng以上DNA(OD260/OD280=1.90,OD260/OD230=2.16),纯度均符合质检标准(1.8

图2 基因组DNA电泳质量检测Fig.2 gDNA electrophoresis
2.2 MspI酶切条件优化
本实验提及的3种RRBS文库构建方法都需要使用MspI酶切富集CpG Island位点,所以需要优化MspI酶切条件,酶切条件选用37 ℃做为酶切温度,选择酶切时间1 h设置酶用量10 U,20 U,40 U,再根据MspI的酶切特性,选择10 U的酶活设置酶切时间条件为0.5 h,1.0 h,2.0 h,4.0 h,O/N(16 h)。通过电泳图(见图3)可以看到,酶用量加大和酶切时间延长并不能让MspI的酶切效率提高,所以从时间和成本的角度出发,酶切条件使用10 U,0.5 h,酶切温度37 ℃,这里需要说明,10 U的酶切用量已经可以将1 μg DNA酶切完整,所以本课题所用100 ng DNA使用10 U,0.5 h,37 ℃的酶切条件完全可以消化完全。
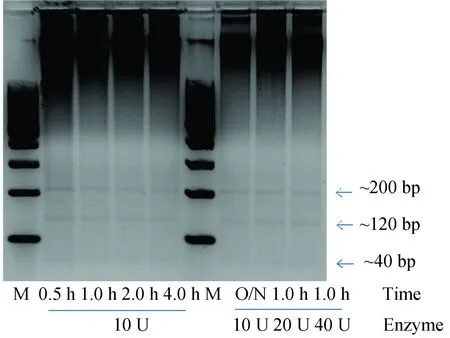
图3 MspI酶切条件优化电泳检测Fig.3 MspI digest condition optimization
2.3 片段大小筛选方法优化
二代测序仪在测序时需要进行成簇反应(Cluster Generarion),成簇反应对DNA片段长度有一定要求,根据Illumina官方解释,正常情况下,成簇反应需求DNA片段长度在200~500 bp之间,所以文库片段过大或过小都会影响成簇反应进行,这也使得筛选合适的DNA片段大小成为文库构建中不可缺少的一步。另外,片段筛选的另一个作用是去除接头二聚体,接头二聚体对测序质量会造成极大影响,据统计,5%的测序接头二聚体含量会导致最终数据50%的损失。片段筛选最常见的方法是琼脂糖凝胶电泳,本课题因起始DNA量很少,导致连接反应的产物在琼脂糖凝胶上基本不能显现,导致切胶时容易误判并且胶回收效率降低,所以本文采用了加入Carrier DNA的方式提高辨识度和胶回收效率,由于在片段大小筛选前,已经使用重亚硫酸盐处理过连接产物,Carrier DNA因不含测序扩增接头序列,在最终的PCR扩增中不会被扩增,所以不会影响最终的测序结果。本实验采用加入Carrier DNA和未加入Carrier DNA进行对照实验,主要比较在琼脂糖凝胶(见图 4(a))上的可辨识度,图上可以很清楚的加入Carrier DNA的目的电泳片段区域(150~350 bp)较未加入Carrier DNA的目的电泳片段区域清晰,切胶时可以明显的将接头二聚体与目的序列分开,有效提高辨识度并且在胶回收过程中,提高了回收效率,且Carrier DNA可以填补硅胶柱中的死体积,从而达到提高回收效率的目的。
使用加入Carrier DNA的方法可以提高胶回收的得率,但微量DNA通过琼脂糖凝胶后的损失率仍然可达到50%以上,为了进一步减少损失,采用了羧基磁珠(Ampure XP Beads)进行文库大小筛选,筛选原理是因羧基磁珠在PEG的影响下,结合DNA的能力会产生变化,PEG含量越高,磁珠可结合的片段就越多,从片段大小上来看,能结合的小片段就越多,使用不同浓度的PEG对打断后的DNA进行筛选,并使用琼脂糖凝胶电泳检测分布(见图4(b)),由图中可以看到,12%~15%的PEG浓度已经可以满足RRBS文库构建的需要,为验证该方案的可重复性,继续设计优化实验如下:取1μg Fragment DNA(200~600 bp),分别用12%/15%筛选和10%/12%筛选,并采用BioAnalyzer进行片段大小分析,做1个重复(见图4(c)),从2 100检测图种可以看到,12%/15%可用于RRBS文库构建中的片段大小筛选,与琼脂糖凝胶分离切胶方法相比,得率和稳定性有较大提高(见图4(c))。
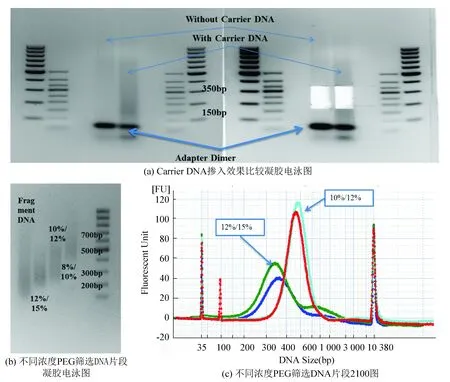
图4 片段大小筛选方法优化Fig.4 Size Selection Optimization
2.4 EA Method 与 Conventional Method比较
EA Method(EA-M)方案主要是将CO-M中的末端修复及3’端加dA合并成一步,并且在做完末端修复和3’端加dA之后,直接加入T4 ligase,减少了其中2步纯化步骤,从而减少DNA损失。该方法可以将末端修复及3’端加dA步骤合并使用了Taq Polymerase在72 ℃下可以在3’端加dA的特性,将Taq Polymerase加入到末端修复的体系中即可实现3’端加dA的目的,并且将这一步的buffer替换成T4 DNA Ligase Buffer,这就使得在进行下一步连接操作时,不需进行纯化。本实验使用100 ng,50 ng,10 ng gDNA,每个实验组设置3个重复,共9个样本,分别使用CO-M和EA-M进行RRBS文库构建,本实验中PCR控制12个循环数,并使用Qubit测定并统计比较2种方法的文库总量(见图5(a)) ,CO-M法在处理10 ng,50 ng gDNA时文库构建失败,在处理100 ng DNA时3个样本的其中1个失败,2个样本得率很低,EA-M法在处理10 ng DNA时得率很低,文库质量差,处理50 ng,100 ng DNA时已能得到很好的结果。
本实验使用1 μg gDNA起始CO-M进行RRBS文库构建作为文库合格的阳性参照,该阳性参照使用Qubit测定文库浓度并计算总量为151.2 ng,并使用BioAnalyzer 2100分别测定阳性文库(见图5(b))和EA-M构建的文库(见图5(c)),使用EA-M法构建的RRBS文库与阳性对照文库相比,大小均分布在200~550之间,没有接头二聚体(Adapter dimer),图中看到BioAnalyzer2100(Agilent)检测出的峰不呈现良好的正态分布,是因RRBS文库构建中DNA片段化使用MspI酶切产生的酶切末端导致,不会对后续测序结果产生任何影响。

图5 EA-M与CO-M构建RRBS文库比较*Fig.5 EA-M vs CO-M
*注:(a)∶ EA-M和CO-M构建RRBS文库,DNA起始分别使用10 ng,50 ng,100 ng,蓝色*标注使用CO-M建库,红色*标注使用EA-M建库;(b)∶ CO-M 文库大小分布;(c)∶ EA-M文库大小分布。
彩图见电子版(http∶//swxxx.alljournals.cn.ch/login.aspx)(2017年第1期doi∶10.3969/j.issn.1672-5565.2017.01.201609001)。
2.5 EA Method稳定性测试
根据EA Method与Conventional Method比较试验的结果,发现EA Method在处理50 ng DNA起始量时,已经有很好的效果。为了进一步测试EA Method的稳定性,将600 ng gDNA平均分成12份(每份50 ng),采用EA Method进行RRBS文库构建,使用Qubit测定文库得率并统计CV(见图 6),发现EA Method在处理50 ng DNA进行RRBS文库构建时,成功率高,文库产出也很稳定(CV<0.05)。
2.6 WB Method与EA Method比较
由上述研究可以看出,EA-M法对于微量样本的处理已初具优势,但因为需要进行切胶和多次DNA转移导致处理50 ng以下的样本仍比较困难,为了能进一步降低DNA出发量和优化文库质量,在磁珠筛选文库大小方案的启发下,建立了全程将DNA与磁珠包被的文库构建方案,该方案的优势在于,DNA全程与磁珠结合,大大减少DNA转移的次数,提高最终有效文库得率。该方案与CO-M法的步骤基本相同,但在所有的酶反应中,DNA都与磁珠结合在一起进行孵育,纯化方案中,采用PEG Buffer进行结合和洗脱,在连接反应前,使用的PEG浓度均为20%,连接反应中采用12%/15%进行洗脱,本实验采用了EA-M法作为对照,分别取10 ng,50 ng,100 ng做2组重复,RRBS文库构建完成后,Qubit进行测定并统计比较2种方法的文库得率(见图7(a)),BioAnalyzer 2100分别随机抽取2种方法中的1个文库进行文库片段大小测定(见图7(b),7(c)),发现WB-M法在处理更微量DNA样本上比EA有更大的改善,基本可以达到3倍左右的文库总量,并且得到的文库范围与EA法有高度一致性(见图7(a)~图7(c))。
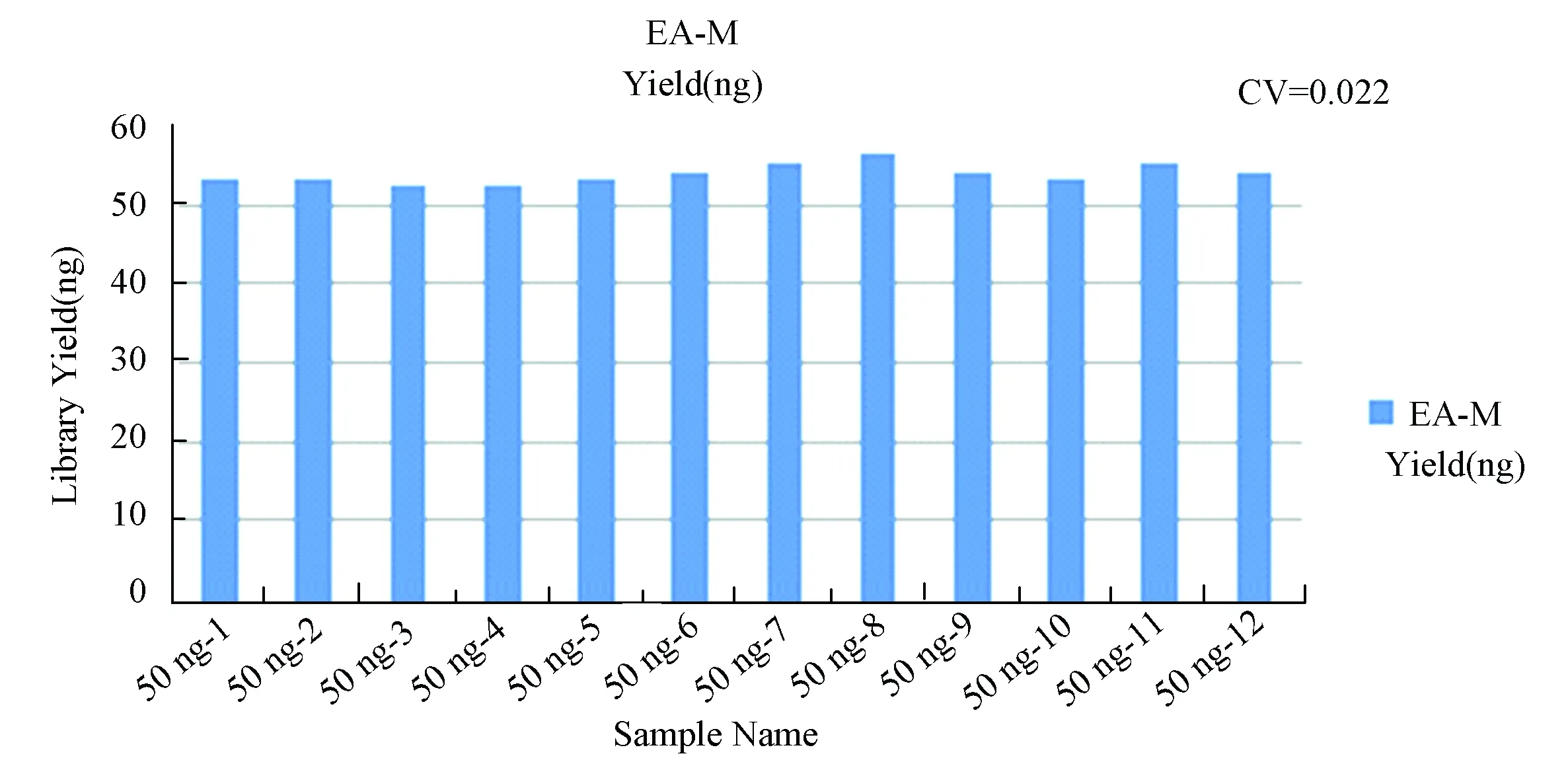
图6 EA方案稳定性测试Fig.6 EA Method stability test
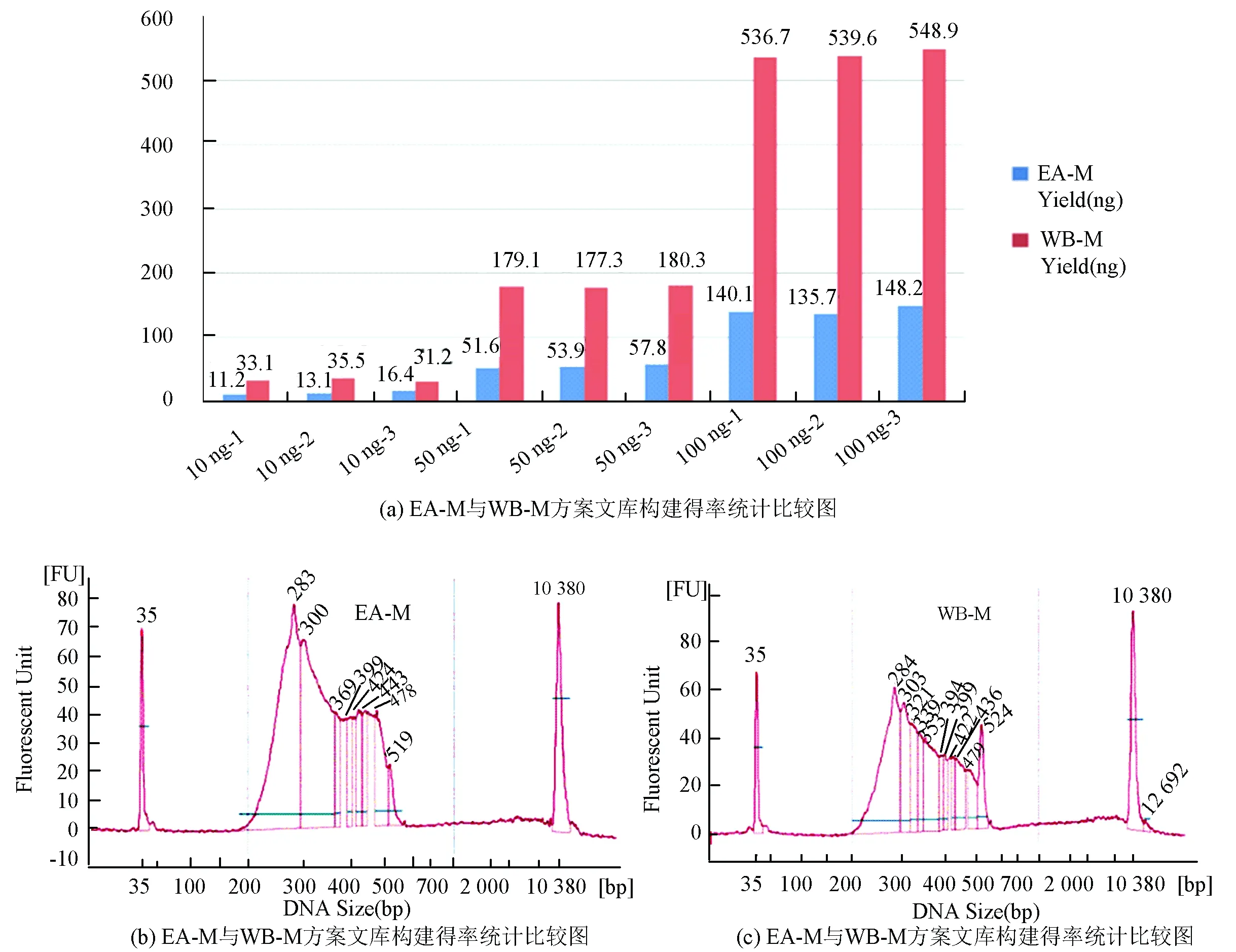
图7 WB-M与EA-M构建RRBS文库比较*Fig.7 WB-M vs EA-M
*注:(a)∶ EA-M和CO-M构建RRBS文库,DNA起始分别使用10 ng,50 ng,100 ng,蓝色*标注使用CO-M建库,红色*标注使用EA-M建库;(b)∶ CO-M 文库大小分布;(c)∶ EA-M文库大小分布。
彩图见电子版(http∶//swxxx.alljournals.cn.ch/login.aspx)(2017年第1期doi∶10.3969/j.issn.1672-5565.2017.01.201609001)。
2.7 WB Method稳定性测试
根据WB Method与EA Method比较试验的结果,发现WB Method在处理10 ng gDNA起始量时,也有较为满意的结果。为了进一步测试WB Method的稳定性,将120 ng gDNA均分成12份样本(每份10 ng),采用WB Method进行RRBS文库构建,使用Qubit测定文库得率并统计CV,发现WB Method在处理10ng DNA进行RRBS文库构建时,成功率高,文库产出也很稳定(CV<0.05)(见图 8)。

图8 WB方案稳定性测试Fig.8 WB Method stability test
2.8 WB-M验证实验
使用WB-M对3个人来源样本分别进行常量(1 μg)RRBS和微量(10 ng)RRBS文库构建,并在Illumina Hiseq3000平台上进行PE150测序,每个样本至少获得20 mol/L Reads数据。数据使用Trim_galore进行数据质量QC(见表1),可见3个常量样本和3个对应微量样本的QC结果一致,得到高质量(Q Score>20)数据后,再使用Bismark将数据比对至人类参考基因组(hg19版本),可看到常量与微量对应样本数据Unique Reads均可达65%(见表1),再将得到的bam文件统计各个位点甲基化位点的甲基化比例及覆盖度,并比较常量和微量样本的差异,可见从不同甲基化水平碱基分布来看,微量和常量样本均符合RRBS文库的特点,甲基化区域均富集在Reads的两头(见图9(c),9(d)),并且覆盖CpG区域2X以上的reads占比80%以上(见图9(a),图9(b)),两者保持了高度一致。最后统计了甲基化通常发生的CpG,CHG,CHH三种不同的context下C碱基的甲基化比例,其中CpG context最常见(见表2),可以看到常量和微量样本的甲基化水平也保持一致。
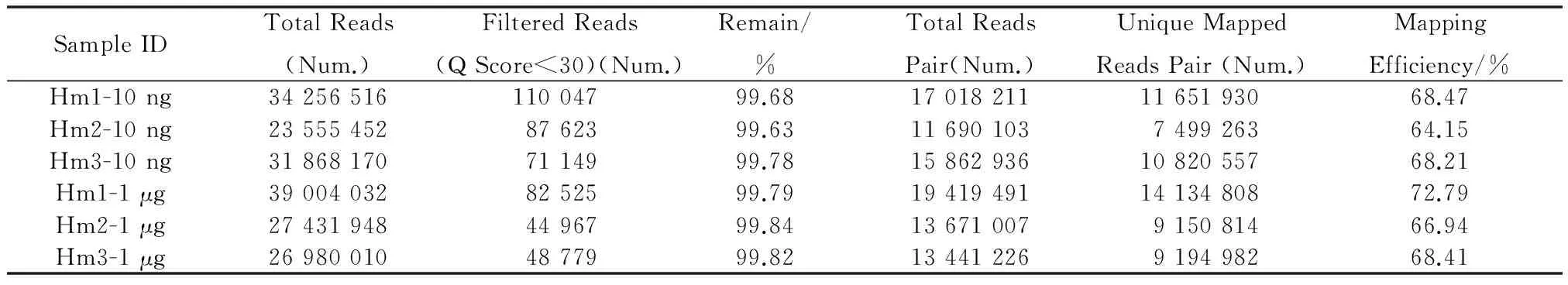
表1 WB方案实验数据QC Table 1 WB Method Data QC Analysis

表2 WB方案CpG,CHG,CHH中C碱基甲基化比例Table 2 WB Method MethyC Content In CpG,CHG,CHH %

图9 WB方案甲基化覆盖度Fig.9 WB method methylation base coverage
3 讨论
随着表观遗传学研究的深入,二代测序在甲基化测序中的研究方法也大幅增多,RRBS(简化甲基化)测序以经济,分辨度高的优点在人、小鼠、大鼠的甲基化研究中被高频使用。RRBS文库是测序前样本处理的关键步骤,其中的核心问题包括:(1)RRBS文库需要符合MspI酶切的特性;(2)RRBS文库必须没有接头二聚体存在(约125~128 bp);(3)文库总量必须满足二代测序基本需求(20 ng以上);(4)实验方法的稳定性(可重复性)高。本研究中使用的2种改进后的基于微量DNA样品的RRBS文库构建方法,可以很好的解决以上4个问题,从而满足RRBS文库用于后续测序的需求。首先,经过MspI酶切优化实验,确定了MspI酶的最优酶切条件,并且在最终文库中也可以明显的看到人gDNA被MspI 酶完整酶切的特征序列,并且文库分布在200~600 bp,有利于在测序仪上进行成簇反应(cluster generation);其次,通过片段大小筛选优化方案,切胶纯化通过加入carrierDNA 提高胶的辨识度,从而将接头二聚体与目的片段间有效分离,磁珠筛选通过不同比例PEG浓度将小片段完整去除,两种方案都可以有效的将接头二聚体去除,保证后期测序数据的高质量;第三,EA-M法通过末端修复以及加“A”反应,连接合并成一步,大大缩短了步骤,从而提高DNA回收效率,实现提高最终文库得率的目的,WB-M法通过将DNA固定在磁珠上的方法减少DNA的被转移次数,从而提高最终文库总量,两种方法分别从减少步骤和减少DNA转移次数的角度提高了文库得率,从而实现10~50 ng微量DNA起始样品的RRBS文库构建;最后,分别通过2种方法的稳定性实验,可以看到2种方法的可重复性很高,成功率基本达到100%,并且针对同一个DNA样本的重复性可以达到CV<0.05的要求,表明这2种方法构建RRBS文库稳定性较好,并且WB-M因全程使用磁珠纯化,适用于液体工作站上使用,在有效提高建库规模化的同时,有效减少人工参与带来的建库误差。
本研究中最关键的因素在于DNA起始量低,所以根据不同的DNA起始量,研发了2种方案,其中EA-M可以针对50~100 ng的微量DNA样品的RRBS文库构建,WB-M可以针对10~50 ng 的微量DNA样品的RRBS文库构建,然而WB-M方案针对50~100 ng DNA出发量时会有更好的效果。原因如下:(1)经过2个方案的比较发现,减少DNA转移次数可以大大降低DNA在建库中的损耗及损伤,DNA被固定在磁珠上对DNA自身也具有一定保护作用,所以WB-M方案可以有效保证酶切末端,减少在建库过程中酶切末端的损伤。(2)Unique Reads Ratio(唯一不重复Reads比例)是二代测序数据质控中的一个重要指标,而影响该指标的因素主要是DNA起始量和文库构建中PCR的循环数。本课题研究的微量DNA RRBS文库构建方案,起始DNA样本量少成为影响该指标的制约因素,因此如何有效减少文库构建中PCR循环数,成为本研究中的关键因素。WB-M可以有效减少DNA损耗,使最终减少PCR循环数变成可能,在后续实验中,发现50 ng DNA起始样本量可以将最终PCR循环次数降低至10次,10 ng DNA起始可有效降低PCR循环数至14次,大大提高测序数据的有效性。经过2种方案的比较发现,WB-M方法在构建微量DNA RRBS文库时,比CO-M和EA-M更加适合,但EA-M在文库构建时间上有一定优势,因为减少了纯化和反应程序,所以EA-M法构建RRBS文库比其他2种方法减少约1/3时间。另一方面,从经济角度来看,EA-M的成本是CO-M的70%左右,WB-M的成本是CO-M的77%左右,所以在进行100 ng 左右DNA起始RRBS文库时,从时间和经济的角度上,推荐使用EA-M法进行文库构建,但从质量上来看,WB-M法则更为优秀。
本研究中因偏重于人来源的微量样本研究(例如:石蜡切片,显微微切割等),所以只使用了人来源的gDNA样本,后期验证WB-M(因WB-M方案制备的文库质量更优于EA-M方案,且在实际应用中WB-M方案更适合检测技术发展方向,所以本验证实验只验证了WB-M方案。)也只使用了人来源样本,并通过信息学分析可看到WB-M在同时处理常量样本和微量样本时,Unique Reads均可达到65%以上,并且可发现的CpG context中C碱基甲基化比例也保持高度一致。本研究中提及的WB-M已成功应用于显微微切割样本的处理。目前还未应用于FFPE样本的处理,从WB-M方案来看,可以适用于FFPE样本的处理,但在进行MspI酶切之后,应使用UDG和FpG酶对FFPE来源DNA进行DNA修复,从而提高文库构建的成功率。随着二代测序技术的发展,单细胞测序也广泛应用到医疗及诊断领域,如PGD,CTCs等,本课题研究中推荐的WB-M方案也可应用于单细胞领域,通过单细胞扩增技术将单个细胞DNA少量扩增后,使用WB-M方案进行微量DNA样品的RRBS文库构建,可以获得高质量的RRBS文库,为后续测序及生物信息分析打下坚实基础。
References)
[1]GOLDBERG A D, ALLIS C D, BERNSTEIN E. Epigenetics:a landscape takes shape[J]. Cell, 2007, 128(4):635-638.DOI:10.1016/j.cell.2007.02.006.
[2]BIRD A P. CpG islands as gene markers in the vertebrate nucleus[J]. Trends in Genetics, 1987, 3(12):342-347.DOI∶10.1016/0168-9525(87)90294-0.
[3]BIRD A. DNA methylation patterns and epigenetic memory[J]. Genes & Development, 2002, 16(1):6-21.DOI:10.1101/gad.947102.
[4]黄庆,郭颖, 府伟灵.人类表观基因组计划[J].生命的化学, 2004, 24 (2):101-103.
HUANG Qing, GUO Yin, FU Weiling.Human epigenome project [J]. Chemistry of Life, 2004,24(2):101-103.
[5]ESTELLER M, ALMOUZNI G. How epigenetics integrates nuclear functions. Workshop on epigenetics and chromatin:transcriptional regμLation and beyond[J]. EMBO Reports, 2005, 6 (7):624-628.DOI:10.1038/sj.embor.7400456.
[6]GARDINER G M, FROMMER M. CpG islands in vertebrate genomes[J]. Journal of MolecμLar Biology, 1987, 196(2): 261-282. DOI∶10.1016/0022-2836(87)90689-9.
[7]TAKAI D, JONES P A. Comprehensive analysis of CpG islands inhuman chromosomes 21 and 22[J]. Proceedings of the National Academy of Sciences of the United States of America, 2002, 99(6):3740-3745.DOI: 10.1073/pnas.052410099.
[8]WANG Y, LEUNG F C. An evaluation of new criteria for CpG islandsin the human genome as gene markers[J]. Bioinformatics, 2004, 20(7):1170-1177.DOI:10.1093/bioinformatics/bth059.
[9]LARSEN F, GUNDERSEN G, LOPEZ R, et al. CpG islands as gene markersin the human genome[J]. Genomics, 1992, 13(4):1095-1107.DOI:10.1016/0888-7543(92)90024-M.
[10]HANSEN R S, STOGER R, WIJMENGA C, et al. Escape from genesilencing in ICF syndrome:evidence for advanced replication timeas a major determinant[J]. Human MolecμLar Genetics, 2000, 9(18):2575-2587.DOI:10.1093/hmg/9.18.2575.
[11]YAMADA Y, WATANABE H, MIURA F, et al. A comprehensive analysisof allelic methylation status of CpG islands on human chromosome21q[J]. Genome Research, 2004, 14(2):247-266.DOI:10.1101/gr.1351604.
[12]HANAHAN D, WEINBERG R A. The hallmarks of cancer[J]. Cell, 2000,100(1):57-70.DOI:10.1016/S0092-8674(00)81683-9.
[13]SuLTAN M,SCHuLZ M H,RICHARD H,et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome[J].Science,2008,321(5891) :956-960.DOI:10.1126/science.1160342.
[14]SCHUSTER S C. Next-generation sequencing transforms today’s biology[J]. Nature Methods,2008,5(1) :16-18.DOI:10.1038/nmeth1156.
[15]COKUS S J, FENG S, ZHANG X, et al.Shotgun bisμLphate sequencing of theArabidopsisgenome reveals DNA methylation patterning[J]. Nature, 2008,452(7184):215-219.DOI:10.1038/nature06745.
[16]LI N,YE M,LI Y,et al. Whole genome DNA methylation analysis based on high throughput sequencing technology[J].Methods,2010,52(3) :203-212.DOI:10.1016/j.ymeth.2010.04.009.
[17]YAN H H, KIKUCHI S, NEUMANN P, et al. Genome-wide mapping of cytosine methylation revealed dynamic DNA methylation patterns associated with genes and centromeres in rice[J]. Plant Journal,2010,63(3) :353-365.DOI:10.1111/j.1365-313X.2010.04246.x.
[18]XIANG H, ZHU J, CHEN Q, et al. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map[J]. Nature Biotechnology, 2010, 28(5) :516-520.DOI:10.1038/nbt.1626.
[19]LISTER R, PELLZZOLA M, DOWEN RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences[J]. Nature, 2009, 462(7271):315-322.DOI:10.1038/nature08514.
[20]TURCOT V, BOUCHARD L, FAUCHER G, et al. DPP4 gene DNA methylation in the omentum is associated with its gene expression and plasma lipid profile in severe obesity[J]. Obesity,2011, 19(2):388-95.DOI:10.1038/oby.2010.198.
[21]LI Y, ZHU J, TIAN G, et al. The DNA methylome of human peripheral blood mononuclear cells[J]. PLoS Biology, 2010,8(11):e1000533.DOI:10.1371/journal.pbio.1000533.
[22]MEISSNER A, GNIRKE A, BELL G W, et al. Reduced representation Bisulfite sequencing for comparative high-resolution DNA methylation analysis[J]. Nucleic Acids Research, 2005,33(18):5868-5877.DOI:10.1093/nar/gki901.
[23]SMITH Z D, GU H C, BOCK C, et al. High-throughput Bisulfite sequencing in mammalian genomes[J]. Methods,2009, 48(3):226-232.DOI:10.1016/j.ymeth.2009.05.003.
[24]GU H C, Bock C, MIKKELSEN T S, et al. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution[J]. Nature Methods, 2010, 7(2):133-136. DOI:10.1038/nmeth.1414.
[25]WANG L, SUN J, WU H, et al. Systematic assessment of reduced representation Bisulfite sequencing to human blood samples:A promising method for large-sample-scale epigenomic studies[J]. Journal of Biotechnology,2012, 157(1):1-6.DOI:10.1016/j.jbiotec.2011.06.034.
[26]WANG J, XIA Y, LI L, et al. Double restriction-enzyme digestion improves the coverage and accuracy of genome-wide CpG methylation profiling by reduced representation Bisulfite sequencing[J]. BMC Genomics,2013, 14(1):1-12.DOI:10.1186/1471-2164-14-11.
[27]CHRISTOPH B, ELENI M T, ARIE B B, et al. Quantitative comparison of genome-wide DNA methylation mapping technologies[J]. Nature biotechnology,2010, 28(10):1106-1114.DOI:10.1038/nbt.1681.
[28]GU H,SMITH Z D,BOCK C, et al. Preparation of reduced representation Bisulfite sequencing librariesfor genome-scale DNA methylation profiling[J]. Nature Protocol, 2011, 6(4):468-481.DOI:10.1038/nprot.2010.190.
[29]MAXIMILIAAN S, ANJA S, LOBS A K, et al. Laser capture microdissection-reduced representation Bisulfite sequencing (LCM-RRBS)maps changes in DNA methylation associated with gonadectomy-induced adrenocortical neoplasia in the mouse[J]. Nucleic Acids Research, 2013, 41(11 ):249-274.DOI:10.1093/nar/gkt230.
An μLtra-low-input RRBS (Reduced Representation Bisulfite Sequencing) library preparation method
WEI Dongkai1,2, NAN Peng2, XU Kangping1, QIU Feng1, 3*
(1.BasePairBio-TechnologyCo.Ltd,JiangSuSuzhou215123,China;2.SchoolofLifeSciencesFudanUniversity,Shanghai200438,China;3.ShanghaicenterforBioinformationtechnology,Shanghai201203,China)
RRBS (Reduced Representation Bisulfite Sequencing) is an effective method for DNA methylation study. Library preparation is one of the most critical steps of RRBS experiments. In conventional RRBS library preparation, it usually needs 1 ug or more starting DNA because of less enzyme digested products and losses in various experiment steps. Meanwhile, many clinical samples from patients, such as frozen puncture tissue, FFPE, microdissection, etc., there is only a limited amount of extracted DNA less than 100 ng, and they don’t fit the requirement of the conventional RRBS library preparation, which greatly limits the application field of RRBS technology. This research studies and designs RRBS library preparation using μLtra-low-input DNA samples (<100 ng), mainly by optimizing MspI digestion condition, DNA fragment size selection, reduction of reaction steps and DNA transfer steps in library preparation, which greatly improve the DNA recovery. We set up two effective μLtra-low-input RRBS library preparation methods (EA-Method and WB-Method). The EA-Method is more cost effective, fast, and efficient than WB-Method when DNA input amount around 100 ng, but the library quality is slightly lower than the WB-Method; The WB-Method has higher quality performance when using 10 ng DNA. In order to test the reliability, we construct 3 groups of RRBS libraries (1ug DNA input and 10ng DNA input) from three volunteer samples (whole blood and mouth swab) by WB-Method. The prepared libraries are sequenced on Illumina Hiseq platform. The compatible resμLts are obtained comparing the μLtra-low-input and conventional RRBS method by analysis of detected C bases in the CpG, CHG, CHH methylation rate. Nevertheless, the μLtra-low-input RRBS method can greatly reduce DNA loss and damage during library preparation, and it will be a wider application of RRBS technology to different type samples and promote broader research field of RRBS technology effectively.
NGS; RRBS; Reduced Representation Bisulfite Sequencing; EA-M; WB-M
2016-09-03;
2016-09-20.
魏冬凯,男,硕士研究生,研究方向:高通量测序技术及应用;E-mail:dkwei@basepair.cn.
*通信作者:裘锋,男,研究员,研究方向:高通量测序及液态活检; E-mail:qiufeng@scbit.org.
10.3969/j.issn.1672-5565.2017.01.201609001
Q78
A
1672-5565(2017)01-033-13
猜你喜欢
太原理工大学学报(2021年6期)2021-11-25 13:33:20
猪业科学(2021年3期)2021-05-21 02:05:36
幽默大师(2020年10期)2020-11-10 09:07:22
中华诗词(2019年1期)2019-11-14 23:33:56
中国海洋大学学报(自然科学版)(2019年7期)2019-05-21 07:26:06
中国海洋大学学报(自然科学版)(2019年7期)2019-01-04 16:33:12
天然产物研究与开发(2018年10期)2018-11-06 07:43:50
猪业科学(2018年4期)2018-05-19 02:04:31
广州化工(2016年11期)2016-09-02 00:42:59
中国医科大学学报(2015年10期)2015-03-01 02:09:55
